Страница 64
Из многочисленных мутаций гемоглобина большинство достаточно редки и лишь немногие из них встречаются чаще других, например HbS, HbC, НЬЕ. Большая часть вариантов гемоглобина (около 350) различается единичными аминокислотными заменами, причиной которых являются генные мутации, связанные с заменой оснований в нуклеотидных последовательностях αили β-глобинового семейства. Многие аминокислотные замены существенно не влияют на функцию гемоглобина и не приводят к патологическим проявлениям. Как правило, это замены в обращенных наружу участках полипептидных цепей тетрамера.
Замены аминокислот, нарушающие нормальную спиральную структуру цепей, часто вызывают неустойчивость гемоглобина. Замена в участках, которыми αи β-цепи контактируют друг с другом, влияют на сродство гемоглобина к кислороду. Нарушения функций гемоглобина, возникающие в результате таких изменений структуры αи β-глобиновых генов, ведут к появлению заболеваний, которые можно разделить на четыре основные группы.
1. Гемолитические анемии. Проявляются в распаде эритроцитов, зависящем от неустойчивости гемоглобина (описано около 100 вариантов нестабильных гемоглобинов с мутациями в гене β-цепи).
2. Метгемоглобинемии. Обусловлены ускоренным окислением двухвалентного железа до трехвалентного и образованием гемоглобина М (известны пять таких мутаций в генах αи β-цепей, состоящих в замене одного основания).
3. Эритроцитоз. Заключается в образовании большего, чем обычно, количества эритроцитов, что обусловлено повышенным сродством гемоглобина к кислороду, который с трудом высвобождается в тканях (таких мутаций известно около 30).
4. Серповидно-клеточная анемия. Заключается в замене гемоглобина НЬА на HbS, который отличается растворимостью и кристаллизацией в условиях гипоксии, что приводит к изменению формы эритроцитов, и проявляется фенотипическим многообразием симптомов (см. рис. 3.21).
Заболевания первых трех групп наследуются по доминантному типу, так что гетерозиготы по мутантному гену страдают нарушением здоровья. Наследование серповидно-клеточной анемии при обычных условиях осуществляется по рецессивному типу, но в условиях сильной гипоксии, например при нахождении на высоте свыше 3000 м над уровнем моря гетерозиготы НbА HbS также страдают анемией.
Описанные мутантные формы гемоглобина возникают в результате изменений структуры генов по типу замены оснований. Мутации иного характера приводят к появлению аллелей глобинов, обусловливающих другие виды патологии. Так, нарушение процесса рекомбинации между аллельными генами (неравноценный кроссинговер) приводит к изменению числа нуклеотидов в них. Следствием этого может быть сдвиг рамки считывания. Нередким результатом таких структурных изменений генов является подавление синтеза той или иной цепи гемоглобина, приводящее к развитию патологических состояний, известных под общим названием талассемии.
Деления одного нуклеотида в 139-м триплете α-глобинового гена, состоящего из 141 триплета, приводит к сдвигу рамки считывания и прочитыванию в новой рамке терминирующего 142-го кодона. При этом (α-глобиновая цепь удлиняется на пять дополнительных аминокислот. Такой особенностью α-цепей характеризуется гемоглобин Vayne. Когда деления располагается ближе к 5'-концу, активный продукт не синтезируется и развиваются различные формы α-, βи γ-талассемий.
Некоторые варианты гемоглобинов возникают в результате дупли-каций. Так, гемоглобин Grady несет дупликацию 116—118 аминокислотных остатков в γ-цепи. В гемоглобине Cranston удлинение р-цепи до 158 аминокислотных остатков является результатом дупликации AG-последовательности после 144-го триплета и последующего сдвига рамки с пропитыванием терминального кодона.
Описанное выше свидетельствует о том, что различные отклонения в структуре ДНК глобиновых генов приводят к замене аминокислот или удлинению полипептидных цепей. Это является причиной образования многих вариантов гемоглобина, которые определяют развитие у человека заболеваний, наследующихся в ряду поколений.
Не меньший интерес представляют механизмы развития различных заболеваний человека, в основе которых лежат мутации генов, приводящие к синтезу белков-ферментов со сниженной активностью или к его подавлению. Это нарушает течение процессов, катализируемых данными ферментами в клетках организма. Примером наследственно детерминированных повреждений метаболизма в организме человека служит фенилкетонурия, развивающаяся вследствие нарушения процессов обмена аминокислоты фенилаланина и накопления в организме токсических промежуточных продуктов.
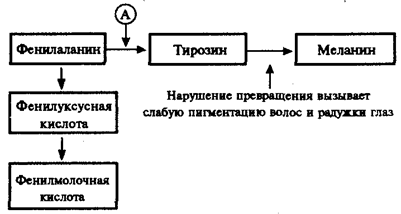
Рис. 4.1. Краткая схема обмена фенилаланина:
А — фермент фенилаланингидроксилаза, наследственный дефект которого приводит к развитию фенилкетонурии
При дефекте фермента фенилаланингидроксилазы фенилаланин не превращается в тирозин (рис. 4.1) и накапливается в крови больных в больших концентрациях (до 0,5—0,6 г/л вместо 0,003— 0,04 г/л в норме). Это приводит к частичному превращению фенилаланина в фенилуксусную и фенилмолочную кислоты, накопление которых наряду с повышенной концентрацией самого фенилаланина оказывает токсическое действие на мозг ребенка. В результате у детей наблюдается различная степень дефекта умственного развития. Нарушение метаболизма фенилаланина сопровождается также нарушением синтеза пигмента меланина, поэтому у больных наблюдается слабая пигментация волос и радужки глаз. Кроме того, высокая концентрация фенилаланина оказывает ингибирующее влияние на ряд ферментных систем, участвующих в превращении других аминокислот: у больных развивается судорожный синдром, нарастает отставание интеллектуального развития. Наследование фенилкетонурии осуществляется по рецессивному типу.
Таким образом, рассмотренные выше примеры демонстрируют весь спектр действия молекулярно-генетических механизмов, обеспечивающих образование в человеческом организме белков как нормально функционирующих, так и обусловливающих развитие различных патологических состояний. Из сказанного по поводу гемоглобина следует, что, во-первых, образование главного функционального белка эритроцитов находится под генным контролем, во-вторых, формирование тетрамерной формы этого белка, с которой связана его физиологическая активность, требует взаимодействия неаллельных генов αи β-глобинов.
Специфический контроль небелковой части молекулы гемоглобина также имеет место и осуществляется независимо, через гены ферментов, необходимых для синтеза гема. Особенности проявления патологических признаков у носителей мутантных аллелей свидетельствуют о существовании определенных отношений между ними и нормальными аллелями. Так, аллель серповидно-клеточности в сочетании с нормальным аллелем (3-глобина (НbА HbS) проявляет себя в обычных условиях как рецессивный. Так же ведет себя мутантный аллель гена, детерминирующего синтез фермента фенилаланингидроксилазы. Проявлением взаимодействия между мутантным и нормальным аллелями по типу доминирования последнего является формирование в организме белка с нормальными свойствами у гетерозигот. Отсутствие нормального аллеля в генотипе организма, гомозиготного по мутантному аллелю, приводит к развитию патологического состояния, обусловленного нарушением функциональной активности соответствующего белка.

Обсуждение Биология Том 1
Комментарии, рецензии и отзывы