2.3. морфология повреждения
2.3. морфология повреждения
Нормальная клетка выполненяет довольно определенное количество функций. На ее структуру и функции влияют определенные генетические программы метаболизма, дифференциров-ки (специализации), ограничивающие влияние соседних клеток и метаболического субстрата. А в совокупности клетки способны удовлетворять физиологические запросы организма — поддерживать нормальный гомеостаз.
Под воздействием некоторых избыточных физиологических, а также патологических стимулов в клетках может развиться процесс адаптации, в результате которого они достигают нового устойчивого состояния, которое позволяет им приспособиться к новым условиям. Если лимиты адаптационного ответа клетки исчерпаны, а адаптация невозможна, наступает повреждение клетки. До определенного предела повреждение клетки обратимо. Однако, если неблагоприятный фактор действует постоянно или его интенсивность очень велика, наступают необратимое повреждение клетки и ее смерть. Например, если в каком-либо сегменте миокарда прекращено кровоснабжение в течение 10— 15 мин, но затем восстановлено, то клетки миокарда претерпевают повреждение, однако могут восстановиться и нормально работать. Если же кровоснабжение не налаживается в течение одного часа, то повреждение миокарда становится необратимым и его клетки в указанном сегменте погибают.
Смерть клетки — конечный результат ее повреждения, наиболее распространенное событие в патологии, сопровождающее существование любого типа клетки, главное следствие ишемии (местное малокровие ткани), инфекции, интоксикации, иммунных реакций. Кроме того, это еще и естественное событие в процессе нормального эмбриогенеза, развития лимфоидной ткани, инволюции органа под действием гормонов, а также желанный результат при радиотерапии и химиотерапии рака.
Существует два типа клеточной смерти — некроз и апоптоз. Некроз клетки — наиболее распространенный тип смерти клетки при экзогенных воздействиях, включая ишемию и химические стимулы. Он проявляется резким набуханием или разрушением клетки, денатурацией и коагуляцией цитоплазматических белков, разрушением клеточных органелл.
Апоптоз служит для нормальной элиминации (устранения) ненужных клеточных популяций в процессе эмбриогенеза и при различных физиологических процессах. Главными морфологическими особенностями апоптоза являются конденсация и фрагментация хроматина. Хотя механизмы некроза и апоптоза различны, между этими процессами много общего.
1. Причины повреждения клеток. Гипоксия является исключительно важной и распространенной причиной повреждения и смерти клеток.
Уменьшение кровоснабжения (ишемия), возникающее при появлении препятствий в артериях, обычно при атеросклерозе или тромбозе, является основной причиной гипоксии. Другой причиной может быть неадекватная оксигенация крови при сердечно-сосудистой недостаточности. Снижение способности крови к транспортировке кислорода, например при анемии и отравлении окисью углерода, является третьей и наиболее редкой причиной гипоксии.
Физические агенты включают механическую травму, чрезмерное снижение или повышение температуры окружающей среды, внезапные колебания атмосферного давления, радиацию и электрический шок.
Химические агенты и лекарства. Даже простые химические соединения, такие как глюкоза и поваренная соль, в гипертонических концентрациях могут вызвать повреждение клеток непосредственно или путем нарушения их электролитного го-меостаза. Даже кислород в высоких концентрациях очень токсичен. Следовые количества веществ, известных как яды, такие как мышьяк, цианиды, соли ртути, могут разрушить достаточно большое количество клеток в течение минут и часов. Разрушительным действием обладают также многие факторы внешней среды: пыль, инсектициды и гербициды; промышленные и природные факторы, например уголь и асбест; социальные факторы: алкоголь, курение и наркотики; высокие дозы лекарств.
Инфекционные агенты включают как субмикроскопические вирусы, так и ленточных червей. К ним относятся риккетсии, бактерии, грибы, а также более высокоорганизованные формы паразитов.
Иммунологические реакции могут защищать организм, а могут вызвать его смерть. Хотя иммунная система защищает организм от воздействия биологических агентов, иммунные реакции могут тем не менее привести к повреждению клеток. Развитие некоторых иммунных реакций лежит в основе так называемых аутоиммунных болезней.
Генетические повреждения клеток могут быть следствием, как правило, врожденных пороков развития, например болезни Дауна. Многие врожденные нарушения метаболизма связаны с энзимопатиями.
Дисбаланс питания нередко является основной причиной повреждения клеток. Дефицит белковой пищи, специфических витаминов остается распространенным явлением во многих странах.
2. Механизмы повреждения клеток. Молекулярные механизмы повреждения клеток, приводящие к их смерти, очень сложны. Так же как существует много причин повреждения клеток, так и нет общего, единого механизма их смерти (по R.S.Cotran, V.Cumar, T.Collins, 1998).
Хотя точку приложения повреждающего агента не всегда удается определить, известны четыре наиболее чувствительные внутриклеточные системы. Во-первых, поддержание целостности клеточных мембран, от чего зависит ионный и осмотический гомеостаз клетки и ее органелл, во-вторых, аэробное дыхание, включающее окислительное фосфорилирование и образование АТФ, в-третьих, синтез ферментов и структурных белков, в-четвертых, сохранение генетического аппарата клетки.
Структурные и биохимические элементы клетки настолько тесно связаны, что повреждение в одном месте приводит к обширным вторичным эффектам. Например, нарушение аэробного дыхания повреждает натриевый насос, который поддерживает ионно-жидкостный баланс, что в свою очередь вызывает нарушение внутриклеточного содержания ионов и воды.
Морфологические изменения выявляются только после того, когда нарушения биологической системы клетки проходят некий критический уровень, причем развитие морфологических признаков смертельного повреждения клетки занимает больше времени, чем появление обратимых изменений. Например, набухание клетки обратимо и может развиться в течение нескольких минут. Однако достоверные светооптические изменения, свидетельствующие о смерти клетки, обнаруживаются в миокарде лишь через 10—12 ч после тотальной ишемии, хотя и известно, что необратимые повреждения наступают уже через 20—60 мин. Естественно, ультраструктурные повреждения будут видны раньше, чем светоопти-ческие.
Реакция клеток на повреждающие воздействия зависит от типа, продолжительности и тяжести последних. Так, малые дозы токсинов или непродолжительная ишемия могут вызвать обратимые изменения, тогда как большие дозы того же токсина и продолжительная ишемия приводят к немедленной гибели клетки или медленному необратимому повреждению, приводящему к клеточной смерти.
Тип, состояние и приспособляемость клетки также влияют на последствия ее повреждения. Для ответа клетки на повреждение важны ее гормональный статус, характер питания и метаболические потребности. Поперечнополосатая мышца голени в покое, например, может обойтись без кровоснабжения, а сердечная мышца — нет. Одни и те же концентрации токсина, например четыреххлористого углерода, могут быть безопасными для одного индивидуума, но приводят к гибели клеток печени у другого, что объясняют содержанием в печени ферментов, расщепляющих четыреххлористый углерод до нетоксичных продуктов.
Механизмы действия многих агентов хорошо известны. Целый ряд токсинов вызывает повреждение клеток, воздействуя на эндогенные субстраты или ферменты. При этом особенно чувствительными становятся гликолиз, цикл лимонной кислоты и окислительное фосфорилирование на внутренних мембранах митохондрий. Цианид, например, инактивирует цитохромокси-дазу, а флуороацетат препятствует реализации цикла лимонной кислоты, что приводит к истощению АТФ. Некоторые анаэробные бактерии, такие как Clostridium perfringens, вырабатывают фосфолипиды, которые атакуют фосфолипиды клеточных мембран.
Наиболее важными для развития повреждения и смерти клетки считают четыре -механизма. Во-первых, в основе повреждения клетки при ишемии лежит отсутствие кислорода. При недостаточном поступлении кислорода в ткани образуются его свободные радикалы, вызывающие перекисное окисление ли-пидов, что оказывает разрушительное действие на клетки.
Во-вторых, особую роль в повреждении клетки имеет нарушение гомеостаза кальция. Свободный кальций присутствует в цитозоле в исключительно низких концентрациях по сравнению с таковым вне клетки. Это состояние поддерживается связанными с клеточной мембраной, энергозависимыми Са2+-, Mg^-АТФазами. Ишемия и некоторые токсины вызывают увеличение концентрации кальция в цитозоле путем его избыточного поступления через плазматическую мембрану и высвобождения из митохондрий и эндоплазматической сети. Повышенное содержание кальция является следствием повышения проницаемости плазмолеммы. Оно ведет к активации ряда ферментов, повреждающих клетку: фосфолипаз (повреждение клеточной мембраны); протеаз (разрушение плазмолеммы и белков цитоскелета), АТФаз (истощение запасов АТФ) и эндонуклеаз (фрагментация хроматина).
В-третьих, потеря митохондриями пиридиннуклеотидов и последующее истощение АТФ, а также снижение синтеза АТФ являются характерными как для ишемического, так и для токсического повреждения клеток. Высокоэнергетические фосфаты в форме АТФ необходимы для многих процессов синтеза и расщепления, происходящих в клетках. К этим процессам относятся мембранный транспорт, синтез белка, липогенез и реакции деацилирования — реацилирования, необходимые для фос-фолипидного обмена (ацилирование — введение в молекулы остатка карбоновых кислот). Имеется достаточно данных о том, что истощение АТФ играет важную роль в потере целостности плазмолеммы, что характерно для смерти клетки.
В-четвертых, ранняя потеря избирательной проницаемости плазматической мембраной — постоянный признак всех видов повреждения клеток. Такие дефекты могут возникать из-за потери АТФ и активации фосфолипаз. Кроме того, плазматическая мембрана может быть повреждена в результате прямого действия некоторых бактериальных токсинов, вирусных белков, компонентов комплемента, веществ из лизированных лимфоцитов (перфорины), а также ряда физических и химических агентов.
3. Виды повреждения клеток. Различают три основных формы повреждения клеток: ишемическое и гипоксическое повреждение; повреждение, вызванное свободными радикалами кислорода; токсическое повреждение.
Ишемическое и гипоксическое повреждение чаще всего связано с окклюзией (закупоркой) артерий. Вначале гипоксия действует на аэробное дыхание клетки — окислительное фосфори-лирование в митохондриях. Так как напряжение кислорода в клетке снижается, прекращается окислительное фосфорилиро-вание, а образование АТФ уменьшается или останавливается. Уменьшение содержания АТФ влияет на многие системы клетки. Сердечная мышца, например, прекращает сокращаться через 60 с после окклюзии коронарной артерии, хотя это и не означает смерти кардиомиоцита. Снижение содержания АТФ в клетке и связанное с ним увеличение АМФ вызывают активацию фосфофруктокиназы и фосфорилазы. В результате происходит усиление анаэробного гликолиза, а поддержание энергетических запасов клетки обеспечивается путем образования АТФ из гликогена. АТФ образуется также анаэробно с помощью креатинкиназы из креатинфосфата. Гликолиз сопровождается аккумуляцией молочной кислоты и неорганического фосфата из-за гидролиза фосфатных эфиров. Все это вызывает снижение внутриклеточного рН. Происходит конденсация ядерного хроматина.
Исчезновение АТФ ведет к острому набуханию (отек) клетки — одному из ранних проявлений ишемического повреждения. Отек клетки связан с нарушением регуляции объема клетки со стороны плазматической мембраны. Клетки млекопитающих обладают высоким внутриклеточным осмотическим коллоидным давлением, на которое влияет концентрация белка, более высокая внутри клеток, а не вне их. Чтобы уравновесить это давление, содержание натрия поддерживается внутри клеток на более низкой концентрации, чем вне клеток. Делается это с помощью энергетически зависимого натриевого насоса, который также поддерживает концентрацию калия внутри клетки значительно более высокой, чем внеклеточная. Нарушение этого активного транспорта ведет к аккумуляция натрия внутри клетки и диффузии калия за ее пределы. Приток жидкости в клетку сопровождается ее набуханием и расширением цистерн эндоплазматической сети. Второй механизм набухания клеток при ишемии — увеличение внутриклеточной осмотической нагрузки, вызванное накоплением катаболитов, таких как неорганические фосфаты, лактат и пуриновые нуклеозиды. Наблюдаются отделение рибосом от мембран шероховатой эндоплазматической сети и диссоциация полисом в моносомы, возможно, из-за разрыва энергетически зависимых связей между мембранами эндоплазматической сети и рибосомами.
Если гипоксия продолжается, развиваются и другие изменения, отражающие повышенную проницаемость клеточной мембраны и ослабление функции митохондрий. На поверхности клеток образуются выпячивания, содержащие только цитозоль, а клетки, имеющие на поверхности микроворсинки, начинают их терять (эпителий проксимальных канальцев почек). В цитоплазме клеток и вне их появляются миелиновые фигуры, формирующиеся из вещества цитоплазмы клеток и мембран органелл. Наблюдается набухание митохондрий, связанное с потерей контроля над их объемом. Цистерны эндоплазматической сети остаются расширенными. Клетки выглядят сильно набухшими, с повышенным содержанием воды, натрия и хлорида натрия, но сниженной концентрацией калия. Все описанные изменения обратимы при условии восстановления снабжения кислородом.
Если же ишемия продолжается, то развиваются необратимые изменения, которые морфологически ассоциируются с выраженной вакуолизацией митохондрий и их крист, повреждением плазматических мембран и набуханием лизосом. В матриксе митохондрий появляются крупные хлопьевидные аморфные уплотнения. В миокарде признаки необратимости повреждения наблюдаются через 30—40 мин после начала ишемии. Массивное поступление кальция в клетку сопровождается потерей белков, ферментов и РНК. Клетки могут также терять метаболиты, необходимые для восстановления запасов АТФ. Повреждение лизосомальных мембран приводит к выделению их ферментов в цитоплазму и активации кислых гидролаз. Так как лизосомы содержат РНКазы, ДНКазы, протеазы, фосфатазы, глюкозидазы и катепсины, происходит расщепление компонентов клетки, которое сопровождается разрушением рибонуклеопротеинов, дез-оксирибонуклеопротеинов и гликогена, а также различными изменениями в ядре.
Вслед за гибелью клетки ее компоненты прогрессивно разрушаются, происходит выброс ферментов клетки во внеклеточное пространство. Наконец, умершие клетки превращаются в образования, состоящие из фосфолипидов в виде миелиновых фигур. Последние подвергаются фагоцитозу и разрушаются до жирных кислот. Проникновение ферментов сквозь поврежденную клеточную мембрану в сыворотку крови позволяет определять параметры смерти клетки клинически. Сердечная мышца, например, содержит трансаминазы, лактатдегидрогеназу и креа-тинкиназу. Повышение содержания этих ферментов в сыворотке крови является клиническим критерием инфаркта миокарда (смерть кардиомиоцитов).
Таким образом, основными признаками необратимости повреждения клетки служат необратимые изменения митохондрий, приводящие к потере АТФ, и плазматических мембран. Основным фактором патогенеза необратимого повреждения клетки при гипоксии является разрушение ее мембраны, в основе которого лежит ряд биохимических механизмов.
Во-первых, в некоторых ишемизированных тканях, например печени, необратимые изменения сопровождаются заметным уменьшением содержания фосфолипилов в клеточной мембране, которое происходит под действием кальцийзависи-мых фосфолипаз. Во-вторых, активация протеаз, связанная с повышением концентрации кальция в цитозоле, ведет к повреждению цитоскелета, выполняющего роль якоря между плазматической мембраной и внутренним содержимым клетки. В результате в процессе набухания клетки происходит отслойка клеточной мембраны от цитоскелета, что делает мембрану более податливой к растяжению и разрыву. В-третьих, при ишемии появляется небольшое количество высокотоксичных свободных радикалов кислорода.
Таким образом, основными причинами гибели клетки при гипоксии являются нарушение окислительного фосфорилиро-вания, приводящее к истощению АТФ и повреждение мембран клетки, а важнейшим медиатором необратимых биохимических и морфологических изменений является кальций.
Повреждение клетки, вызванное свободными радикалами кислорода, чаще всего возникает под воздействием химических
веществ, ионизирующего излучения, кислорода и других газов, при старении клеток, разрушении опухолей макрофагами и в некоторых иных случаях.
Свободные радикалы представляют собой молекулы кислорода, имеющие один непарный электрон на внешней орбите. В таком состоянии радикал исключительно активен и нестабилен и вступает в реакции с неорганическими и органическими соединениями — белками, липидами, углеводами и особенно с ключевыми молекулами в мембранах и нуклеиновых кислотах. Более того, свободные радикалы инициируют аутокаталитичес-кие реакции, в ходе которых молекулы, с которыми они реагируют, также превращаются в свободные радикалы, вызывающие цепь разрушений.
Образование свободных радикалов в клетках может быть инициировано ультрафиолетовыми и рентгеновскими лучами, эндогенными окислительными реакциями, связанными с нормальным метаболизмом клетки, а также происходит при ферментном расщеплении некоторых химических веществ и лекарств, поступающих в клетку извне. Непарный электрон может быть связан практически с любым атомом, но в биологических объектах — чаще всего с кислородом, углеродом и азотом. В норме под воздействием цитохромоксидазы кислород превращается в воду. Однако в клетке образуются и токсичные промежуточные соединения кислорода. Наиболее важными среди них являются супероксид, перекись водорода и гипероксильные ионы. Они образуются под влиянием различных окислительных ферментов в цитозоле, митохондриях, лизосомах, пероксисомах и плазматической мембране.
Перекисное окисление липидов мембран. Свободные радикалы в присутствии кислорода могут вызывать перекисное окисление липидов плазматической мембраны и мембран органелл. Липидно-радикальные взаимодействия приводят к образованию перекисей, которые сами по себе являются активными соединениями, инициирующими последующее повреждение других жирных кислот. Таким образом возникает цепь ауто-каталитических реакций, приводящая к обширному повреждению клеток.
Окислительное превращение белков. Свободные радикалы вызывают перекисное связывание таких лабильных аминокислот, как метионин, гистидин, цистин и лизин, а также фрагментацию полипептидных цепей. Окислительное превращение усиливает разрушение ключевых ферментов посредством нейтральных протеаз, содержащихся в цитозоле.
Повреждение ДНК. Свободные радикалы вступают в реакцию с тимином, входящим в состав ДНК, что приводит к гибели клетки или ее злокачественному превращению.
Конечный результат действия свободных радикалов зависит от баланса между их образованием и разрушением.
3. Пальцев, т. 1.
65
Токсическое повреждение возникает под действием химических веществ, которые воздействуют на клетки посредством одного из двух механизмов. Во-первых, часть водорастворимых соединений может действовать непосредственно, связываясь с некоторыми молекулами или органеллами. Например, при попадании в организм хлорида ртути происходит связывание сульфгидрильных групп клеточной мембраны, которое приводит к повышению проницаемости последней и торможению АТФаза-зависимого транспорта. В подобных случаях наиболее выраженные изменения наблюдаются в клетках, которые используют, абсорбируют, выделяют или концентрируют эти химические соединения. Поэтому при попадании в организм хлорида ртути в наибольшей степени страдают клетки желудочно-кишечного тракта и почек. Цианид действует непосредственно на ферменты митохондрий. Многие противоопухолевые химио-терапевтические препараты, в том числе антибиотики, также вызывают повреждение клеток благодаря их цитотоксическому действию.
Во-вторых, большинство химических соединений, особенно жирорастворимые токсины, биологически неактивно и вначале превращается в токсичные метаболиты, которые затем действуют на клетки-мишени. Хотя эти метаболиты могут вызывать повреждение мембран и клеток путем прямого ковалентного связывания с мембранными белками и липидами, наиболее важным является образование свободных радикалов кислорода и последующее перекисное окисление липидов.
4. Морфология повреждения и смерти клеток. Обратимые повреждения. В классической морфологии нелетальное повреждение клеток называется дистрофией, или обратимым повреждением клеток. Светооптически различают два вида таких изменений: набухание и жировые изменения.
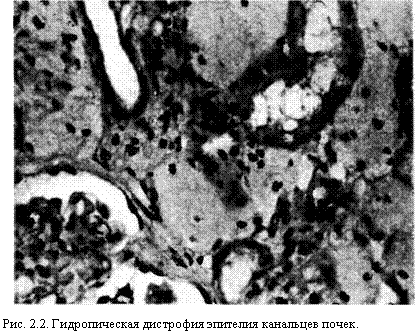
Набухание развивается тогда, когда клетки не способны поддерживать ионный и жидкостный гомеостаз (рис. 2.2).
Жировые изменения также могут быть признаком обратимого повреждения клетки. Для них характерно появление мелких или крупных липидных включений в цитоплазме. Они встречаются при гипоксических й различных формах токсических повреждений, главным образом в клетках, участвующих или зависящих от обмена жиров, таких как гепатоциты (рис. 2.3, А и Б) и миокардиоциты (рис. 2.4).
Некроз наряду с апоптозом является одним из двух морфологических выражений смерти клетки и представляет собой спектр морфологических изменений, которые развиваются вслед за смертью клетки в живой ткани. Это результат разрушающего действия ферментов на летально поврежденную клетку. Фактически развиваются два конкурирующих процесса: ферментное переваривание клетки и денатурация белков. Каталитические ферменты выходят из лизосом гибнущей клетки (аутолиз) или из лизосом
 лейкоцитов (гетеролиз). В зависимости от того, происходит денатурация белков или ферментное переваривание, развивается одна из двух разновидностей некроза. В первом случае наблюдается коагуляционный некроз, а во втором — колликвационный (разжижающий) некроз.
лейкоцитов (гетеролиз). В зависимости от того, происходит денатурация белков или ферментное переваривание, развивается одна из двух разновидностей некроза. В первом случае наблюдается коагуляционный некроз, а во втором — колликвационный (разжижающий) некроз.
Требуется несколько часов для развития обоих процессов, и поэтому в случае внезапной смерти, вызванной, например, инфарктом миокарда, соответствующие структурные изменения в пораженных миокардиоцитах просто не успевают развиться. Наиболее ранние гистологические признаки некроза сердечной мышцы появляются через 10—12, в лучшем случае через 8 ч после включения механизмов необратимой гибели ткани (рис. 2.5). Однако утрата ферментов, например лактатдегидрогеназы в зоне поражения, может быть надежно определена с помощью реакции с трифенилтетразолием уже через 4—6 ч. Как макро-, так и микроскопически зона инфаркта элективно окрашивается в темно-красный цвет.
На ранних этапах некротических изменений в ткани может развиваться эозинофилия цитоплазмы погибающих клеток. Это связано с утратой нормальной слабо выраженной ба-зофилии, обеспечиваемой в цитоплазме РНК, а также с повышением оксифильности клеточных белков, подвергающихся все возрастающей денатурации. По мере ферментного пере-
з*
67
Рис. 2.3, А. Жировая инфильтрация печени (окраска гематоксилином и эозином).
варивания органелл в цитоплазме на их месте появляются вакуоли. Однако все эти процессы могут лишь косвенно говорить о развивающемся некрозе. Да они и не столь постоянны.
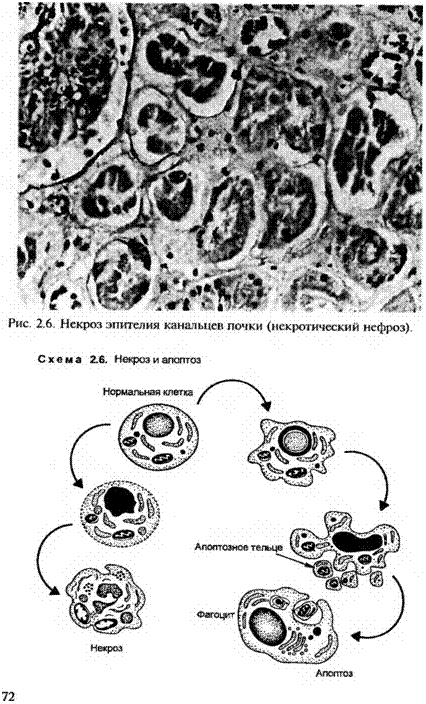
Куда более надежными признаками патологической гибели ткани являются изменения ядер клеток. Так, в результате, по-видимому, активности ДНКаз исчезают не только базофилия хроматина, но и контуры, и вообще какие-либо субстраты ядер (рис. 2.6). Это кариолизис — наиболее достоверный признак некроза. Другим таким маркерным изменением является карио-пикноз — сморщивание и гиперхромность ядра, вызванные конденсацией ДНК, третьим признаком служит кариорексис — фрагментация или разрыв ядра на глыбки. Скопление последних в некротическом поле называется детритом, или частицами детрита (кашицеобразной массой распада ткани). Массы некро-тизированных клеток формируют очаг некроза.
Различают пять видов некроза: коагуляционный, колликва-ционный, гангренозный (гангрена), казеозный и жировой.
Коагуляционный некроз подразумевает сохранение общих контуров очага в течение по крайней мере нескольких дней. Предполагается, что само повреждение или возрастающий впоследствии внутриклеточный ацидоз денатурирует не только
структурные белки, но и ферменты и тем самым блокирует протеолиз клетки. Коагуляционный некроз характерен для ги-поксической гибели ткани во всех органах, кроме головного мозга. Типичный пример — инфаркт миокарда. С течением времени некротизированные миокардиоциты фрагментиру-ются и удаляются с помощью фагоцитов (макрофагов и лейкоцитов) и протеолиза лизосомальными ферментами лейкоцитов.
Колликвационный (влажный) некроз развивается в результате аутолиза или гетеролиза (аутолиз — распад клеток под влиянием разных ферментов). Чаще он встречается в очагах поражений бактериальными инфекционными агентами и обусловлен разжижающим действием лейкоцитарных ферментов в этих очагах. Что касается влажного некроза при гипоксической гибели ткани головного мозга, то причины его развития остаются неясными. Некоторые исследователи объясняют природу такого некроза тем, что ткань мозга богата водой и процессы аутолиза в ней преобладают над коагуляционными изменениями.
Гангрена — некроз черного или очень темного цвета, развивающийся в тканях, прямо или через анатомические каналы соприкасающихся с внешней средой. Помимо конечностей, гангрена возникает в легких, кишечнике, коже щек и других мес
тах. При сухой гангрене некроз имеет коагуляционный характер. Влажная гангрена развивается при инфицировании погибшей ткани бактериями, обычно анаэробными, например из группы клостридий. Темный цвет гангренозной ткани создается сульфидом железа, образующимся из железа гемоглобина и сероводорода воздуха. Разновидностью сухой или влажной гангрены является пролежень. Изредка встречается газовая гангрена, при которой пузырьки с сероводородом, произведенным обычно микробом Clostridium welchii, находятся внутри некротизиро-ванной ткани.
Казеозный (творожистый, сыровидный) некроз как частная разновидность коагуляционного чаще всего появляется в туберкулезных очагах. Макроскопически он действительно напоминает творог или мягкий сыр. Микроскопически для него характерна гранулематозная реакция, представленная туберкулезными бугорками (см. главу 14).
Жировой (ферментный) некроз, или стеатонекроз, представляет собой очаги разрушенной жировой клетчатки замазкооб-разного вида разной формы и величины. Чаще всего это следствие освобождения активированных липаз поджелудочной железы, действующих прямо в брюшной полости при остром панкреатите. У живых людей большинство некротизированных клеток и их остатков исчезает в результате комбинированного проРис. 2.5. Исчезновение гликогена из мышечных клеток в зоне ишемии миокарда (ШИК-реакция).
цесса ферментного переваривания и фрагментации и последующего фагоцитоза остатков клеток лейкоцитами. Если же клетки и их остатки полностью не разрушаются и не реабсорбируются, они подвергаются кальцификации (дистрофическое обызвествление).
Если некроз считается патологической формой клеточной смерти, возникающей в результате резкого повреждающего воздействия на клетку, то апоптоз противопоставляется ему как контролируемый процесс самоуничтожения клетки.
Подробные данные об ультраструктурных нарушениях в клетках при апоптозе и некрозе получены в результате электронно-микроскопических и биохимических исследований. При некрозе на ранних стадиях наблюдается конденсация хроматина, затем происходит набухание клетки с разрушением ци-топлазматических структур и последующим лизисом ядра. Морфологическими проявлениями апоптоза являются конденсация ядерного гетерохроматина и сморщивание клетки с сохранением целостности органелл. Клетка распадается на апоптозные тельца, представляющие собой мембранные структуры с заключенными внутри органеллами и частицами ядра, затем апоптозные тельца фагоцитируются и разрушаются при помощи лизо-сом окружающими клетками (схема 2.6).
При некрозе в результате повреждающего воздействия на клетку резко уменьшается синтез макроэргических фосфатов с последующим нарушением проницаемости мембран и их целостности. Внутриклеточные антигены попадают в межклеточное пространство и индуцируют воспалительный ответ.
При апоптозе повреждение ДНК, недостаток факторов роста, воздействие на рецепторы, нарушение метаболизма ведут к активации внутренней программы самоуничтожения. Синхронно с уплотнением хроматина под влиянием эндонуклеаз начинается деградация ДНК до фрагментов в 180—200 пар оснований. Показано, что эндонуклеазы расщепляют двойную цепочку ДНК между нуклеосомами. В результате активации цитоплазматических протеаз происходит разрушение цито-скелета. межклеточных контактов, связывание белков и распад клетки на апоптозные тельца. Быстрое распознавание и фагоцитоз апоптозных телец указывают на наличие на их поверхности специфических рецепторов, облегчающих адгезию и фагоцитоз. Важнейшим свойством апо-птоза считается сохранение внутриклеточного содержимого в мембранных структурах, что позволяет осуществить элиминацию клетки без развития воспалительного ответа.
Характерные признаки апоптоза связаны с характером воздействия и типом клеток.
Конденсация хроматина связана с расщеплением ядерной ДНК, которое происходит в участках связей между нуклеосомами и приводит к образованию фрагментов. Такие фрагменты создают характерную для апоптоза картину ядра в отличие от некроза, при котором ядро выглядит пятнистым. Эта интернуклеосомальная фрагментация ДНК развивается с участием кальцийчувствительной эндонуклеазы. Эндонуклеаза постоянно присутствует в некоторых типах клеток, например ти-моцитах, в других клетках фермент образуется перед началом апоптоза.
Нарушения объема и размеров клеток связывают с активностью трансглутаминазы. Этот фермент вызывает перекрестное связывание цитоплазматических белков, образующих оболочку под плазматической мембраной.
Фагоцитоз апоптозных телец макрофагами и другими типами клеток обеспечивается рецепторами последних. Одним из таких рецепторов у макрофагов является витронекти-новый рецептор оц,рЗ-интегрин, обеспечивающий фагоцитоз апоптозных нейтрофилов.
Одной из важных особенностей апоптоза является его з а-висимость от активации генов и синтеза белка. Индукция апоптозспецифических генов обеспечивается за счет специальных стимулов, таких как протеины теплового шока и протоонкогены.
Некоторые гены, участвующие в возникновении и росте злокачественной опухоли (онкогены и супрессорные гены), играют регуляторную роль в индукции апоптоза. Например, онкоген р53 стимулирует апоптоз в норме.
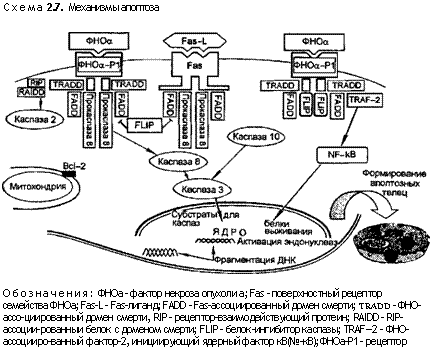 Механизм запуска апоптоза и внутриклеточной передачи сигнала от рецепторной системы, приводящий к запуску генетической программы самоуничтожения клетки, связан с системой рецепторов ФНОа (ФНОа-RiyFas (схема 2.7). Fas является трансмембранным белком, принадлежащим к семейству рецепторов ФНОа. Рецептор к Fas — Fas-L (Fas-ligand), который называется также ФНОр, индуцирует апоптоз клеток при взаимодействии с Fas — Fas-Ь/ФНОа-Ю -ФНОа. Взаимодействие Fas и Fas-L или ФНОа-Rl и ФНОа вызывает образование индуцирующего смерть клетки белкового комплекса — DISC (death inducing signaling complex). При образовании комплекса Fas — Fas-L в процесс вовлекается белок-адаптер: Fas и Fas-ассоциированный домен смерти — FADD (Fas-associated death domain), который связывается с соответствующим доменом смерти цитоплазматической части Fas. Затем FADD взаимодействует с проэнзимной формой фермента каспазы-8, вызывая димеризацию так называемого эффекторного домена смерти каспазы-8. В формирование белкового комплекса может быть вовлечен рецепторвзаимодействующий протеин — RIP (receptor-interacting protein). Описаны и другие молекулы, такие как RAIDD (RIP associated ICH/CED-3-homologous protein with death domain) и CRADD (caspase and RIP adapter with death domain), участвующие в этом сигнальном пути и индуцирующие апоптоз. Взаимодействие ФНОа с ФНОа-Rl вызывает образование DISC, которое включает в себя активацию каспазы-8, RIP и FADD, как и при образовании комплекса Fas — Fas-L. Кроме того, в ассоциации ФНОа с ФНОа-Rl участвует специфический белок — ФНО-ассоциированный домен смерти — TRADD (TNF-associated death domain).
Механизм запуска апоптоза и внутриклеточной передачи сигнала от рецепторной системы, приводящий к запуску генетической программы самоуничтожения клетки, связан с системой рецепторов ФНОа (ФНОа-RiyFas (схема 2.7). Fas является трансмембранным белком, принадлежащим к семейству рецепторов ФНОа. Рецептор к Fas — Fas-L (Fas-ligand), который называется также ФНОр, индуцирует апоптоз клеток при взаимодействии с Fas — Fas-Ь/ФНОа-Ю -ФНОа. Взаимодействие Fas и Fas-L или ФНОа-Rl и ФНОа вызывает образование индуцирующего смерть клетки белкового комплекса — DISC (death inducing signaling complex). При образовании комплекса Fas — Fas-L в процесс вовлекается белок-адаптер: Fas и Fas-ассоциированный домен смерти — FADD (Fas-associated death domain), который связывается с соответствующим доменом смерти цитоплазматической части Fas. Затем FADD взаимодействует с проэнзимной формой фермента каспазы-8, вызывая димеризацию так называемого эффекторного домена смерти каспазы-8. В формирование белкового комплекса может быть вовлечен рецепторвзаимодействующий протеин — RIP (receptor-interacting protein). Описаны и другие молекулы, такие как RAIDD (RIP associated ICH/CED-3-homologous protein with death domain) и CRADD (caspase and RIP adapter with death domain), участвующие в этом сигнальном пути и индуцирующие апоптоз. Взаимодействие ФНОа с ФНОа-Rl вызывает образование DISC, которое включает в себя активацию каспазы-8, RIP и FADD, как и при образовании комплекса Fas — Fas-L. Кроме того, в ассоциации ФНОа с ФНОа-Rl участвует специфический белок — ФНО-ассоциированный домен смерти — TRADD (TNF-associated death domain).
В обеих рецепторных системах в результате формирования DISC активируется каспаза-8, которая в свою очередь активирует каскад других белков этого семейства с передачей сигнала на ядро и индукцией апоптоза. Однако некоторые вирусы и клетки могут содержать специфические белки — FLIPs (FLICE inhibitory proteins) — ингибиторы каспазы-8.
Изучение роли цистеиновых протеаз в осуществлении апоптоза началось с обнаружения специфических генов клеточной гибели на генетической модели — нематоде Caenorhabditis ele-gans. У животных, несущих мутацию в специфических ced-генах (от англ. cells death — клеточная смерть), процесс клеточной гибели блокируется на определенных стадиях. Показано, что экспрессия генов ced-З и ced-4 инициирует смерть для всех типов соматических клеток нематоды. Если один из этих генов инак-тивирован, все клетки, погибающие в норме в течение эмбрионального развития, выживают. Оказалось, что ген ced-З кодирует у нематод белок, сходный с белками группы цистеиновых протеаз млекопитающих, к которой принадлежит ИЛ-^-конвертирующий фермент — ICE (interleukin-ip-converting enzyme). Эти исследования позволяют предположить, что основные генетические механизмы регуляции апоптоза сохранены в процессе эволюции и примерно одинаковы для всех животных, от нематод до млекопитающих. Позднее были открыты другие цистеи-новые протеазы, и это семейство получило название каспазы (caspases — cysteinyl aspartate-specific proteases). Помимо каспазы-8, в настоящее время предполагается участие в апоптозе кас-паз 2, 3, 6, 7, 9, 10. Субстратами для каспаз служат ядерные и сигнальные белки, например ДНК-зависимая протеинкиназа и поли(АДФ-рибоза)полимераза.
На той же модели С. elegans идентифицирован другой ген, участвующий в клеточной смерти, — ced-9. Функцией этого гена является защита клетки от возможной смерти. Установлено, что этот ген кодирует белок, аналогичный Всі-2 млекопитающих (см. главу 7). Ген Bcl-2 (B-cell lymphoma-2) был впервые описан при фолликулярных В-клеточных лимфомах человека, несколько позже была установлена его апоптозная активность в отсутствие факторов роста. В настоящее время, помимо Вс1-2, известны и другие члены этого семейства. Bcl-2, Bcl-XL, Bcl-w и некоторые другие ингибируют апоптоз, а Вах, Bik, Bad, Bid, Bcl-Xs способствуют его индукции. Эти белки в разных сочетаниях служат агонистами и антагонистами друг другу, образуя гомои гетеродимеры. Протеины семейства Вс1-2 локализуются на мембранах митохондрий, эндоплазматической сети и ядра. Механизмы действия Вс1-2, вероятно, связаны с регуляцией транспорта Са2+ и взаимодействием с каспазами.
В некоторых тканях Fas-опосредованный апоптоз может быть вызван только при одновременном действии блокатора белкового синтеза циклогексимида. Блокада Fas-опосредованного апоптоза в этих тканях возможна за счет активации NF-kB (nuclear factor — кВ)-опосредованной транскрипции. Взаимодействие ФНО с ФНОа-Rl вызывает активацию TRAF-2 (TNF associated factor-2), который индуцирует образование активной формы NF-kB, ответственной за усиление транскрипции анти-апоптозных белков. Помимо семейства Вс1-2, другие онкогены также участвуют в регуляции апоптоза.
Онкоген р53 играет важную роль в управлении клеточным циклом, подавляя его до начала репликации ДНК. При повреждении ДНК повышенный синтез р53 вызывает апоптоз клетки. Однако, по всей вероятности, этот ген не участвует в апоптозе, вызванном другими факторами.
Недостаточно ясна роль онкогена с-тус в регуляции апоптоза. Первоначально было установлено, что с-тус является необходимым фактором для вступления клеток в клеточный цикл. Однако впоследствии оказалось, что в определенных условиях с-тус может индуцировать апоптоз. В настоящее время считается, что с-тус участвует в регуляции как процесса пролиферации, так и апоптоза, взаимодействуя с другими онкогенами, такими как р53 и bcl-2.
Апоптоз ответствен за многочисленные физиологические и патологические процессы, идущие в организме:
апоптоз опосредует запрограммированное удаление клеток в процессе эмбриогенеза (включая имплантацию, органогенез и инволюцию):
гормонзависимая инволюция клеток у взрослых, например отторжение клеток эндометрия в процессе менструального цикла, атрезия фолликулов в яичниках во время менопаузы, регрессия лактирующей грудной железы после прекращения кормления ребенка регулируются с помощью апоптоза;
уничтожение клеток в пролиферирующих клеточных популяциях, таких как эпителий крипт тонкой кишки;
смерть клеток в опухолях;
смерть аутореактивных клонов Т-лимфоцитов;
патологическая атрофия гормонзависимых тканей, например атрофия простаты после кастрации и исчезновение лимфоцитов в тимусе после введения гликопротеидов;
патологическая атрофия паренхиматозных органов после перекрытия протока, например поджелудочной железы, околоушной слюнной железы, почки;
смерть клеток, вызванная цитотоксическими Т-клетками, например, при отторжении трансплантата;
гибель клеток при некоторых вирусных заболеваниях, например при вирусном гепатите, при котором фрагменты клеток при апоптозе известны как тельца Каунсилмена (WT. Councilman);
клеточная смерть, вызванная различными слабыми повреждающими воздействиями, которые в больших дозах приводят к гибели клетки (термальные воздействия, радиация, цитотоксические противоопухолевые препараты и, возможно, гипоксия).
5. Субклеточные изменения при повреждении клеток (по R S.Cotran, V.Kumar, T.Collins, 1998)
Гетерофагия и аутофагия. Лизосомы содержат различные гидролитические ферменты, в том числе кислую фосфатазу, глюкуронидазу, сульфатазу, рибонуклеазу, коллагеназу и др. Эти ферменты синтезируются в шероховатой эндоплазматичес-кой сети, а затем упаковываются в аппарате Гольджи. На этой стадии их называют первичными лизосомами. Первичные лизосомы сливаются с окруженными мембраной вакуолями, содержащими продукты переваривания, и образуют фаголизосомы. Лизосомы участвуют в утилизации фагоцитированного материала посредством гетерофагии и аутофагии.
Гетерофагия представляет собой феномен, посредством которого материал извне захватывается клеткой с помощью эндоцитоза. Поглощение частиц называется фагоцитозом, а растворенных мелких макромолекул — пиноцитозом. Гетерофагия характерна для фагоцитирующих клеток, таких как нейтрофилы и макрофаги. В качестве примеров гетерофагоцитоза можно привести поглощение бактерий нейтрофильными лейкоцитами и удаление апоптозных клеток и телец макрофагами. Слияние фагоцитарной вакуоли с лизосомой заканчивается растворением захваченного материала.
При аутофагии внутриклеточные органеллы и порции цитозоля вначале отделяются от цитоплазмы в аутофагические вакуоли, образованные из свободных от рибосом мембран шероховатой эндоплазматической сети, которые затем сливаются с первичными лизосомами или элементами комплекса Гольджи, образуя аутофаголизосому. Аутофагия — распространенный феномен, направленный на удаление разрушенных органелл поврежденной клетки. Он особенно выражен в клетках, атрофирующихся в результате недостаточного питания или гормональной инволюции.
Ферменты лизосом способны разрушать большинство белков и углеводов, хотя некоторые липиды все равно остаются непереваренными. Лизосомы с непереваренными остатками встречаются в клетках в виде остаточных телец. Гранулы пигмента липофусцина представляют собой непереваренный материал, который образовался после внутриклеточного перекисно-го окисления липидов. Некоторые нерастворимые пигменты, такие как частицы угля, попадающие из атмосферы, или пигмент, вводимый при татуировке, могут находиться в фаголизо-сомах макрофагов десятилетиями.
В лизосомах накапливаются также вещества, которые клетки не могут адекватно метаболизировать. При болезнях накопления, для которых характерен дефицит ферментов, разрушающих макромолекулы, происходит ненормальное накопление этих веществ в лизосомах клеток всего тела, особенно в нейронах, приводя к развитию тяжелых заболеваний.
Дисфункция митохондрий играет важную роль при остром повреждении клетки. Различные изменения количества, размеров и формы митохондрий наблюдаются в патологических условиях. Например, при гипертрофии и атрофии наблюдаются увеличение и уменьшение количества митохондрий соответственно. Митохондрии могут быть очень крупными и принимать различную форму (мегамитохондрии), например, при алкогольной болезни печени. При некоторых врожденных метаболических заболеваниях скелетных мышц — митохондри-альных миопатиях — дефекты метаболизма митохондрий сочетаются с увеличением их количества, причем митохондрии часто бывают необычно крупными, имеют аномальные кристы и содержат кристаллоиды. Кроме того, некоторые опухоли (слюнных желез, щитовидной и околощитовидной желез, почек), так называемые онкоцитомы, состоят из клеток с множеством вытянутых митохондрий.
Аномалии цитоскелета встречаются при различных заболеваниях. Как уже говорилось, в норме цитоскелет состоит из микротрубочек, тонких актиновых нитей, толстых мио-зиновых нитей и различных промежуточных филаментов. Аномалии цитоскелета делятся на дефекты функций клетки (локомоторная и движение внутриклеточных органелл) и накопления фибриллярного материала внутри клетки.
Функционирующие миофиламенты и микротрубочки необходимы для различных стадий миграции лейкоцитов и фагоцитоза. Поэтому именно с недостаточностью цитоскелета связаны некоторые дефекты движения лейкоцитов в ответ на повреждающие стимулы или неспособность таких клеток осуществлять адекватный фагоцитоз. Например, дефект полимеризации микротрубочек при синдроме Чедиака—Хигаши (A.M.Chediak, O.Higashi) вызывает замедленное слияние лизосом с фагосома-ми в лейкоцитах и, таким образом, нарушается фагоцитоз бактерий, а в цитоплазме лейкоцитов появляются крупные аномальные лизосомы. Некоторые лекарства, такие как цитохала-зин В, тормозят функцию микрофиламентов и, таким образом, нарушают фагоцитоз. Дефекты в организации микротрубочек могут тормозить подвижность сперматозоидов, вызывая стерильность у мужчин, а также приводить к неподвижности ресничек дыхательного эпителия, что препятствует очищению дыхательных путей от бактерий и способствует развитию бронхо-эктазов.
При некоторых типах повреждений клеток наблюдается накопление промежуточных филаментов. Например, тельца Мал-лори (F.B.Mallory), или алкогольный гиалин, представляют собой эозинофильные включения в клетках печени, которые характерны для алкогольной болезни. Такие включения состоят главным образом из промежуточных филаментов. Нейрофиб-риллярные включения в мозге при болезни Альцгеймера (A.Alzheimer) содержат белки и нейрофиламенты и отражают повреждение цитоскелета нейронов.
6. Внутриклеточные скопления. Одним из клеточных проявлений метаболических нарушений является аккумуляция ненормальных количеств различных веществ: 1) воды, липидов, белков и углеводов; 2) аномальных веществ, в том числе экзогенных, таких как ионы, продукты нарушенного метаболизма; 3) пигментов. Все они могут накапливаться транзитно или постоянно и быть безвредными или очень токсичными. Вещества могут быть локализованы в цитоплазме (чаще всего в лизосомах) или в ядре.
Различают три разновидности внутриклеточных скоплений. Во-первых, скопления естественных эндогенных метаболитов, которые образуются в нормальном или ускоренном ритме, а скорость их удаления недостаточна, например при жировых изменениях печени, обусловленных внутриклеточным накоплением триглицеридов (см. рис. 2.3).
Во-вторых, нормальное или ненормальное накопление эндогенных веществ, которые не могут метаболизироваться. Они откладываются внутри клетки в виде аморфных или филамен-тозных скоплений. Важной причиной этого является генетический дефект фермента. В результате некоторые продукты метаболизма не могут быть использованы клеткой и развиваются болезни накопления.
В-третьих, аккумуляция аномальных экзогенных веществ, которые клетка не может ни разрушить с помощью ферментов, ни транспортировать в другое место. Примерами такого типа повреждений клеток служат отложения частиц угля, кремнезема, а также вирусные включения.
Л и п и д ы. Все классы липидов могут накапливаться в клетках: триглицериды, эфиры холестерина и фосфолипиды. Фосфолипиды также входят в состав миелиновых фигур, обнаруживаемых в некротизированных клетках. Аномальные комплексы липидов и углеводов аккумулируются при некоторых генетических болезнях накопления, таких как мукополисахаридо-зы и болезнь Гоше (Ph.Ch.E.Gaucher).
Стеатоз — накопление триглииеридов в паренхиматозных клетках. Чаще всего такие жировые изменения встречаются в печени, которая является главным органом, участвующим в метаболизме жиров, а также в сердце, мышцах и почках. Стеатоз вызывают токсины, сахарный диабет, тучность и другие причины. Одной из наиболее частых причин стеатоза печени является прием алкоголя
Липиды транспортируются в печень из жировой ткани в виде свободных жирных кислот или из пищи в виде хиломикро-нов или свободных жирных кислот. Свободные жирные кислоты попадают в печеночные клетки и превращаются в триглице-риды. Некоторые из них превращаются в холестерол и инкорпорируются в фосфолипиды или окисляются в митохондриях в кетоновые тела. Некоторые жирные кислоты синтезируются непосредственно в печени из ацетата. Прежде чем покинуть печень, внутриклеточные триглицериды соединяются с молекулами апопротеина (липидный акцепторный белок), образуя липопротеины.
Накопление триглицеридов в печени происходит в результате дефектов в процессе превращения жирных кислот в липопро-теин. Возникновение ряда таких дефектов вызывает алкоголь, повреждающий функции митохондрий и микросом. СС14 снижает синтез липидного акцепторного белка. Гипоксия тормозит окисление жирных кислот. Голодание увеличивает мобилизацию жировой ткани и ускоряет синтез триглицеридов.
Значение стеатоза зависит от причины и выраженности накопления липидов. Слабо выраженное накопление липидов не влияет на функцию клеток, а выраженная аккумуляция липидов может нарушать функцию клетки и необратимо повреждать внутриклеточные процессы.
Холестерин и его эфиры. Большинство клеток использует холестерин для синтеза клеточных мембран. Однако при некоторых патологических процессах может происходить накопление холестерина в клетках.
При атеросклерозе холестерин и его эфиры находят в атеро-склеротических бляшках, гладкомышечных клетках и макрофагах в интиме аорты и крупных артерии Некоторые из этих клеток разрываются, выделяя липиды во внеклеточное пространство. Внеклеточный холестерин может кристаллизоваться, приобретая форму длинных игл.
При врожденных гиперлипидемических состояниях скопления пенистых клеток, содержащих холестерин, обнаруживаются в субэпидермальной соединительной ткани кожи и в сухожилиях. Они образуют опухолеподобные скопления (ксан-томы).
Пенистые макрофаги часто встречаются в местах повреждения клеток в очагах воспаления. Они образуются вследствие фагоцитоза холестерина из мембран разрушенных клеток.
Белки. Избыток белка является причиной морфологически выявляемых скоплений вещества в клетках, которые выглядят как округлые эозинофильные капли, вакуоли или массы.
При почечных заболеваниях, связанных с потерей белка с мочой (протеинурия), белки проходят через гломерулярный фильтр в проксимальные канальцы, откуда белок реабсорбиру-ется эпителиальными клетками с помощью пиноцитоза. Пино-цитозные пузырьки сливаются с лизосомами, формируя фаго-лизосомы, которые образуют капли в цитоплазме эпителиальных клеток проксимальных канальцев.
Эндоплазматическая сеть плазматических клеток участвует в активном синтезе иммуноглобулинов и может выглядеть растянутой и заполненной крупными гомогенными эозинофильными включениями, которые называют тельцами Русселя (W.Russell).
Гликоген. Внутриклеточные скопления гликогена встречаются при нарушениях метаболизма глюкозы или гликогена. Например, при сахарном диабете гликоген обнаруживается в эпителиальных клетках дистального отдела извитых канальцев почек и иногда в нисходящей части петли Генле, а также в клетках печени, В-клетках островкового аппарата и кардиомио-цитах. Гликоген также накапливается в клетках при гликогено-зах (болезнях накопления).
Пигменты — окрашенные вещества; одни из них встречаются в клетках в норме, а другие накапливаются при определенных патологических состояниях. Пигменты могут быть экзогенными и эндогенными. Самым распространенным экзогенным пигментом является уголь. Попадая в легкие, он захватывается альвеолярными макрофагами и по лимфатическим каналам проникает в регионарные лимфатические узлы. При татуировках пигмент фагоцитируется макрофагами дермы, в которых и остается.
К эндогенным пигментам относятся липофусцин, меланин и некоторые производные гемоглобина.
Липофусцин — нерастворимый пигмент, известный также как липохром, или пигмент старения. Липофусцин состоит из полимеров липидов и фосфолипидов, связанных с белком. Полагают, что это продукт перекисного окисления полиненасыщенных липидов субклеточных мембран. Его находят в клетках, подвергающихся медленным, регрессивным изменениям, чаще всего в печени и миокарде старых людей или у больных с тяжелыми нарушениями питания или раковым истощением. Обычно происходит уплотнение органа и уменьшение его размеров (бурая атрофия).
Меланин образуется в меланоцитах тогда, когда тирозиназа катализирует окисление тирозина в дегидроксифенилаланин.
Гемосидерин — пигмент гемоглобина, аккумулирующий железо в клетках. В норме железо доставляется с помощью транспортных белков трансферринов. В клетках железо накапливает
ся, связываясь с белком апоферритином и образуя мицеллы ферритина. Ферритин встречается во многих типах клеток в норме. Когда имеется общий или местный избыток железа, ферритин образует гранулы гемосидерина. Таким образом, ге-мосидерин представляет собой агрегаты мицелл ферритина. В норме малые количества гемосидерина встречаются в моно-нуклеарных фагоцитах костного мозга, селезенки и печени — органах, участвующих в разрушении эритроцитов.
Местное накопление железа и гемосидерина бывает при обширных или множестве мелких кровоизлияний. Лучшим примером местного гемосидероза является обычный синяк.
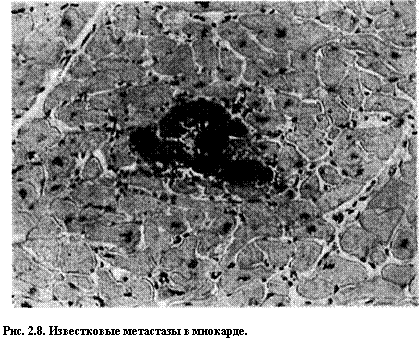
При системном накоплении железа гемосидерин откладывается во многих органах и тканях (гемосидероз), обычно при повышенном всасывании железа из пищи, нарушенной утилизации железа, гемолитических анемиях и переливаниях крови (рис. 2.7).
В большинстве случаев системного гемосидероза пигмент не повреждает паренхиматозные клетки и не вызывает нарушений функций органа. Более выраженное накопление железа, например, при гемохроматозе, сопровождается повреждением печени и поджелудочной железы, развитием фиброза печени, сердечной недостаточности и сахарного диабета.
Билирубин — нормальный пигмент желчи. Он образуется из гемоглобина, но не содержит железа. Билирубин выявляется морфологически в клетках и тканях только при желтухе. Хотя пигмент попадает во все органы, ткани и жидкости тела, его накопление происходит лишь в печени и почках. В печени, особенно при заболеваниях, сопровождающихся нарушением оттока желчи, билирубин встречается в синусоидах, купферовских клетках и гепатоцитах. Скопления билирубина могут вызывать некроз гепатоцитов. Билирубин накапливается также в эпителиальных клетках канальцев почек при различных формах желтухи.
7. Патологическое обызвествление. При патологическом обызвествлении происходит аномальное выпадение солей кальция одновременно с малыми количествами железа, магния и других минеральных солей. Различают две формы патологического обызвествления: дистрофическое и метастатическое.
При дистрофическом обызвествлении соли кальция откладываются в участках некроза. Кальцификация происходит в атеромах при выраженном атеросклерозе, который сопровождается повреждением интимы аорты и крупных артерий. При старении и ревматических болезнях соли кальция откладываются в клапанах сердца
При дистрофическом обызвествлении образуются кристаллические минералы, состоящие из фосфата кальция, в виде апатита, похожего на гидроксиапатит костей. Процесс дистрофического обызвествления складывается из двух фаз: инициации (нуклеации) и распространения — и развивается как в клетках, так и внеклеточно. Вне клеток фаза инициации проходит в окруженных мембраной пузырьках около 200 нм в диаметре. Полагают, что кальцит концентрируется в этих пузырьках благодаря его сродству с кислыми фосфолипидами, а фосфаты аккумулируются, видимо, в результате действия фосфатаз, связанных с мембраной. Инициация внутриклеточного обызвествления происходит в митохондриях умерших или умирающих клеток, которые накапливают кальций.
В фазе распространения происходит образование кристаллов, которое зависит от концентрации кальция и фосфора во внеклеточных пространствах, а также от наличия минеральных ингибиторов, коллагена и других белков. Кроме того, остеопонтин, кислый, связанный с кальцием фосфопроте-ин с высоким сродством к гидроксиапатиту, участвует в минерализации костей и, видимо, в дистрофическом обызвествлении, по крайней мере артерий и почки. А коллаген ускоряет образование кристаллов.
Хотя дистрофическое обызвествление может быть просто признаком повреждения клеток, оно может вызывать и нарушения функций органов, например, при обызвествлении клапанов сердца и атеросклерозе.
 Метастатическое обызвествление происходит в нормальных тканях при гиперкальциемии. Гиперкальциемия усиливает дистрофическое обызвествление. Причиной гиперкальциемии являются гиперпаратиреоидизм, интоксикация витамином D, системный саркоидоз, гипертиреоидизм, идиопатическая гиперкальциемия, болезнь Аддисона (адренокортикальная недостаточность), усиленное разрушение костей, связанное с диссеминированной костной опухолью (множественная миелома и метастатический рак), лейкоз, сниженное образование кости при иммобилизации. Гиперкальциемия в некоторых случаях развивается также при выраженной почечной недостаточности с задержкой фосфора, приводящей к вторичному гиперпаратиреоидизму.
Метастатическое обызвествление происходит в нормальных тканях при гиперкальциемии. Гиперкальциемия усиливает дистрофическое обызвествление. Причиной гиперкальциемии являются гиперпаратиреоидизм, интоксикация витамином D, системный саркоидоз, гипертиреоидизм, идиопатическая гиперкальциемия, болезнь Аддисона (адренокортикальная недостаточность), усиленное разрушение костей, связанное с диссеминированной костной опухолью (множественная миелома и метастатический рак), лейкоз, сниженное образование кости при иммобилизации. Гиперкальциемия в некоторых случаях развивается также при выраженной почечной недостаточности с задержкой фосфора, приводящей к вторичному гиперпаратиреоидизму.
Метастатическое обызвествление может распространяться по всему телу, но обычно поражает интерстициальные ткани кровеносных сосудов, почек, легких и слизистой оболочки желудка (рис. 2.8). Во всех этих участках морфологически соли кальция напоминают таковые при дистрофическом обызвествлении. Таким образом, они могут быть представлены как некристаллическими аморфными депозитами, так и гидроксиапа-титовыми кристаллами. Метастатическое обызвествление обычно начинается в митохондриях, за исключением почечных канальцев, где оно развивается в базальных мембранах, возможно, в связи с внеклеточными пузырьками, отпочковывающимися от эпителиальных клеток.
Чаще всего минеральные соли не вызывают дисфункции органов клинически, однако массивное обызвествление, например, тканей легкого, вызывает выраженную дыхательную недостаточность. Массивные депозиты в почках (нефрокальциноз) могут привести к их повреждению.
8. Гиалиновые изменения. Термин «гиалин» чаще используется как описательная гистологическая характеристика, чем специфический маркер клеточного повреждения. Обычно это повреждение клеток и внеклеточного вещества, которое дает гомогенное розовое окрашивание при использовании гематоксилина и эозина. В качестве примера внутриклеточных гиалиновых депозитов можно привести накопления белка, описанные ранее (капли при реабсорбции в канальцах почки, тельца Руссе-ля и алкогольный гиалин Маллори).
Внеклеточный гиалин труднее поддается анализу. Например, может гиалинизироваться соединительная ткань в старых рубцах.чПри длительной гипертензии и сахарном диабете стенки артерий, особенно почек, становятся гиалинизированными благодаря проникновению белков сквозь стенки сосудов и их отложению в базальной мембране.
Таким образом, хотя термин «гиалин» используется достаточно часто, важно учитывать механизмы, вызывающие эти изменения при различных патологических состояниях.

Обсуждение Патологическая анатомия
Комментарии, рецензии и отзывы