5.2. патология иммунной системы
5.2. патология иммунной системы
Различают четыре основных типа патологических состояний иммунной системы: 1) реакции гиперчувствительности, которые представляют собой иммунологическое повреждение тканей; 2) аутоиммунные болезни, являющиеся иммунными реакциями против собственного организма; 3) синдромы иммунного дефицита, возникающие вследствие врожденного или приобретенного дефекта нормального иммунного ответа; 4) амилоидоз.
Иммунное повреждение тканей (реакции гиперчувствительности) (по R.S.Cotran, V.Kumar, T.Collins, 1998). Контакт организма с антигеном приводит не только к развитию защитного иммунного ответа, но и к появлению реакций, повреждающих ткани. Экзогенные антигены содержатся в пыли, пыльце растений, еде, лекарствах, микробах, химических веществах, во многих препаратах крови, используемых в клинической практике. Иммунные ответы на такие антигены возникают как в форме тривиального дискомфорта (кожный зуд), так и тяжелых заболеваний (бронхиальная астма). Реакции гиперчувствительности могут быть инициированы взаимодействием антигена с антителом или клеточными иммунными механизмами. Иммунные реакции, повреждающие ткани, могут быть связаны не только с экзогенными, но и эндогенными антигенами.
Болезни гиперчувствительности классифицируют на основе иммунологических механизмов, их вызывающих. При I типе реакций гиперчувствительности иммунный ответ сопровождается высвобождением вазоактивных и спазмогенных веществ, при II типе — антитела участвуют в повреждении клеток, делая их восприимчивыми к фагоцитозу или лизису; при III типе —имму-нокомплексных болезнях — взаимодействие антител с антигенами приводит к образованию иммунных комплексов, активирующих комплемент. Фракции комплемента привлекают нейтрофилы, которые вызывают повреждение ткани. При IV типе реакций гиперчувствительности развивается клеточный иммунный ответ с участием сенсибилизированных лимфоцитов.
Реакции гиперчувствительности I типа (анафилактический тип) могут быть местными или системными. Системная реакция развивается в ответ на внутривенное введение антигена, к которому организм хозяина предварительно сенсибилизирован. Местные реакции зависят от места проникновения антигена и имеют характер ограниченного отека кожи (кожная аллергия, крапивница), выделений из носа и конъюнктив (аллергический ринит, конъюнктивит), сенной лихорадки, бронхиальной астмы или аллергического гастроэнтерита (пищевая аллергия).
Реакции гиперчувствительности I типа проходят в своем развитии две фазы. Фаза инициального ответа развивается через 5—30 мин после контакта с аллергеном и характеризуется расширением сосудов, повышением их проницаемости, а также спазмом гладкой мускулатуры или секрецией желез. Поздняя фаза наблюдается через 2—8 ч без дополнительных контактов с антигеном, продолжается несколько дней и характеризуется интенсивной инфильтрацией тканей эозинофилами, нейтрофила-ми, базофилами и моноцитами, а также повреждением эпителиальных клеток слизистых оболочек.
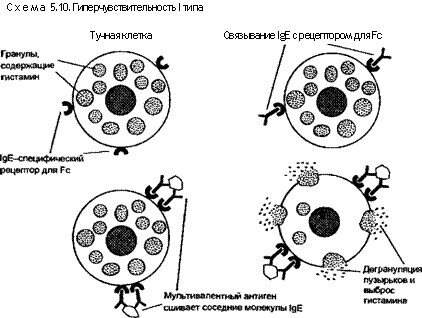 Развитие гиперчувствительности 1 типа у человека обеспечивает IgE. IgE-антитела, образованные в ответ на аллерген, атакуют тучные клетки и базофилы, обладающие высокочувствительными Fc-рецепторами (схема 5.10). При повторном контакте тучных клеток и базофилов, сенсибилизированных цитофильными IgE-антителами, со специфическим антигеном происходит выброс медиаторов, обусловливающих клинические проявления.
Развитие гиперчувствительности 1 типа у человека обеспечивает IgE. IgE-антитела, образованные в ответ на аллерген, атакуют тучные клетки и базофилы, обладающие высокочувствительными Fc-рецепторами (схема 5.10). При повторном контакте тучных клеток и базофилов, сенсибилизированных цитофильными IgE-антителами, со специфическим антигеном происходит выброс медиаторов, обусловливающих клинические проявления.
Тучные клетки и базофилы играют главную роль в развитии гиперчувствительности I типа. Тучные клетки обнаруживаются преимущественно вокруг кровеносных сосудов, нервов и в субэпителиальных областях, где чаше всего развивается реакция гиперчувствительности I типа. Цитоплазма тучных клеток содержит окруженные мембраной гранулы, заполненные биологически активными медиаторами (см. главу 4). Сенсибилизированные Fc-фрагментом IgE тучные клетки и базофилы активируют компоненты комплемента СЗа и С5а (анафилотоксины). Секрецию тучных клеток стимулируют также цитокины макрофагов (ИЛ-8), некоторые лекарства (кодеин, морфин) и физические воздействия (тепло, холод, солнечный свет). Базофилы похожи на тучные клетки из-за наличия у них рецепторов для Fc-фрагмента IgE, а также гранул в цитоплазме. В противоположность тучным клеткам в норме базофилы в тканях не встречаются, а обнаруживаются в небольших количествах лишь в кровотоке.
Связывание молекул IgE инициирует дегрануляцию тучных клеток с выбросом первичных медиаторов, а также синтез de novo и выброс таких вторичных медиаторов, как метаболиты арахидоновой кислоты. С этими медиаторами связано появление новых симптомов гиперчувствительности I типа.
Первичные медиаторы содержатся в гранулах тучных клеток и базофилов и подразделяются на четыре категории. Биогенные амины включают в себя гистамин и аденозин. Гистамин вызывает выраженный спазм гладкой мускулатуры бронхов, усиление сосудистой проницаемости, интенсивную секрецию носовых, бронхиальных и желудочных желез. Аденозин стимулирует тучные клетки к выбросу медиаторов, вызывающих бронхоспазм и торможение агрегации тромбоцитов. К медиаторам хемотаксиса относятся эозинофильный и нейтрофильный хемотакеичес-кие факторы. Ферменты содержатся в матриксе гранул и представлены протеазами (химаза, триптаза), а также некоторыми кислыми гидролазами. Ферменты вызывают образование кини-нов и активацию анафилотоксина СЗа, воздействуя на их предшественников. Протеогликаном является гепарин.
Вторичные медиаторы подразделяются на два класса соединений — липидные медиаторы и цитокины. Липидные медиаторы вызывают активацию фосфолипазы А2 и образование арахидоновой кислоты, из которой в свою очередь возникают лейко-триены и простагландины. Лейкотриены С4и D4 — самые сильные из известных вазоактивных и спазмогенных агентов. Они в несколько тысяч раз сильнее гистамина и вызывают повышение сосудистой проницаемости и сокращение гладкой мускулатуры бронхов. Лейкотриен В4 оказывает хемотаксическое действие на нейтрофилы, эозинофилы и моноциты. Простагландин D2 вызывает сильный бронхоспазм и повышенную секрецию слизи. Фактор активации тромбоцитов (ФАТ) — вторичный медиатор, вызывающий агрегацию тромбоцитов, выброс гистамина, бронхоспазм, повышение сосудистой проницаемости и расширение кровеносных сосудов (см. главу 4). Тучные клетки также продуцируют ряд цитокинов, включая ФНОа, ИЛ-1, ИЛ-2, ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-6 и колониестимулирующий фактор гранулоцитов — макрофагов (КСФ-Г,-М).
Таким образом, гистамин и лейкотриены быстро выделяются из сенсибилизированных тучных клеток и базофилов, обеспечивая немедленно развивающуюся реакцию, характеризующуюся отеком слизистой оболочки, секрецией слизи, спазмом гладкой мускулатуры. Многие другие медиаторы, представленные ФАТ и ФНОа, включаются в позднюю фазу ответа. Среди клеток, которые появляются в позднюю фазу реакции, особенно важны эозинофилы. Набор их медиаторов так же обширен, как и в тучных клетках. Кроме того, они продуцируют главный основной белок (МВР) и эозинофильный катионный белок (ЕСР), которые токсичны для эпителиальных клеток.
Системная анафилаксия возникает после введения гетероло-гичных белков — антисывороток, гормонов, ферментов, полисахаридов, некоторых лекарств, например пенициллина. Тяжесть состояния зависит от уровня предварительной сенсибилизации. Шоковая доза антигена, однако, может быть исключительно мала. Так, для кожного тестирования различных форм аллергии достаточно ничтожных количеств антигена. Через несколько минут после контакта с антигеном появляются зуд, крапивница и кожная, эритема (см. главу 25), затем через короткое время развивается спазм респираторных бронхиол. Рвота, спазмы в животе, понос и отек гортани могут закончиться шоком и смертью больного. На вскрытии у одних больных обнаруживаются отек и кровоизлияния в легких, у других — острая эмфизема легких с дилатацией правого желудочка сердца.
Местную анафилаксию иногда называют атопической аллергией. Около 10 \% населения страдает от местной анафилаксии, возникающей в ответ на попадание в организм аллергенов: пыльцы растений, перхоти животных, домашней пыли и т.д. К заболеваниям, в основе которых лежит местная анафилаксия, относят крапивницу, ангионевротический отек, аллергический ринит (сенную лихорадку) и некоторые формы астмы. Существует семейная предрасположенность к этому типу аллергии.
Реакции гиперчувствительности II типа. При таких реакциях в организме появляются антитела, направленные против собственных тканей, выступающих в роли антигенов. Антигенные детерминанты могут быть связаны с плазмо-леммой или представляют собой экзогенный антиген, адсорбированный на поверхности клетки. В любом случае реакция гиперчувствительности возникает как следствие связывания антител с нормальными или поврежденными структурами клетки. Известны три антителозависимых механизма развития реакций этого типа.
Комплементзависимые реакции. Существуют два механизма, с помощью которых антитело и комплемент могут вызывать гиперчувствительность II типа: прямой лизис и опсонизация. В первом случае антитело (IgM или IgG) реагирует с антигеном на поверхности клетки, активируя систему комплемента (см. главу 4). Оно приводит в действие мембрано-атакующий комплекс (МАК), который нарушает целостность мембраны, продырявливая липид-ный слой. Во втором случае клетки фагоцитируются после фиксации антитела или компонента комплемента СЗЬ к поверхности клетки (опсонизация). При этом варианте гиперчувствительности II типа чаще всего в качестве мишени выступают клетки крови (эритроциты, лейкоциты, тромбоциты), но антитела могут быть направлены также против внеклеточных структур, например против гломерулярной базальной мембраны.
Клинически такие реакции возникают в следующих случаях: при переливании несовместимой крови; эритробластозе плода и антигенных различиях между матерью и плодом, когда антитела (IgG) матери, проникая сквозь плаценту, вызывают разрушение эритроцитов плода; при аутоиммунной гемолитической анемии, агранулоцитозе и тромбоцитопении, когда происходит образование антител против собственных клеток крови, которые затем разрушаются; при отдельных реакциях на лекарства, когда образующиеся антитела реагируют с препаратами и формируют комплексы с эритроцитарным антигеном.
Антителозависимая клеточная цитотоксичность не сопровождается фиксацией комплемента, однако вызывает кооперацию лейкоцитов. Клетки-мишени, покрытые IgG-антителами в низких концентрациях, уничтожаются несенсибилизированными клетками, обладающими Fc-рецепторами. Эти клетки связывают клетки-мишени с помощью рецепторов для Fc-фрагмента IgG, а лизис клеток происходит без фагоцитоза. В этом виде ци-тотоксичности участвуют моноциты, нейтрофилы, эозинофилы и NK-клетки. Несмотря на то что в большинстве случаев в этом типе реакции участвуют антитела IgG, иногда, например при связанной с эозинофилами реакции цитотоксичности против паразитов, задействованы IgE-антитела. Этот вид цитотоксичности имеет значение при реакции отторжения трансплантата.
Антителоопосредованная дисфункция клеток. В некоторых случаях антитела, направленные против рецепторов на поверхности клеток, нарушают их функционирование, не вызывая повреждения клеток или развития воспаления. Например, при миастении антитела вступают в реакцию с ацетилхолиновыми рецепторами в двигательных концевых пластинках скелетных мышц, нарушая нервно-мышечную передачу и вызывая таким образом мышечную слабость. При болезни Грейвса (R.J.Graves) антитела против рецепторов тиреоидстимулирующего гормона стимулируют эпителиальные клетки щитовидной железы, что приводит к гипертиреозу.
Реакции гиперчувствительности III типа (связанные с иммунными комплексами). Развитие таких реакций обусловлено наличием комплексов антиген — антитело, образующихся в результате связывания антигена с антителом в кровеносном русле (циркулирующие иммунные комплексы) или вне сосудов (иммунные комплексы in situ). Циркулирующие иммунные комплексы (ЦИК) вызывают повреждение при попадании в стенку кровеносных сосудов или в фильтрующие структуры (гломерулярный фильтр в почках). Известны два типа иммунокомплексных повреждений, которые формируются при поступлении в организм экзогенного антигена (чужеродный белок, бактерия, вирус) и при образовании антител против собственных антигенов. Заболевания, обусловленные наличием иммунных комплексов, могут быть генерализованными, если эти комплексы образуются в крови и оседают во многих органах, или связанными с отдельными органами, такими как почки (гломерулонефрит), суставы (артрит) или мелкие кровеносные сосуды кожи — местная реакция Артюса (N.M.Arthus).
1. Системная иммунокомплексная болезнь. Одной из ее разновидностей является острая сывороточная болезнь, возникающая в результате многократного введения больших доз чужеродной сыворотки крови для пассивной иммунизации.
Патогенез системной иммунокомплексной болезни складывается из трех фаз: образования в крови комплексов антиген — антитело; осаждения иммунных комплексов в различных тканях; воспалительной реакции. Первая фаза начинается с попадания антигена в кровь и образования антител. Приблизительно через 5 дней после введения сыворотки образуются антитела против ее компонентов, которые, еще находясь в кровотоке, образуют комплексы антиген — антитело. Во вторую фазу эти комплексы оседают в различных тканях. Дальнейшее течение болезни определяют два фактора: размеры иммунных комплексов и состояние системы мононуклеарных фагоцитов (СМФ). При значительном избытке антител образуются очень крупные комплексы, которые быстро удаляются из кровотока клетками СМФ и относительно безвредны. Наиболее патогенны комплексы мелких и средних размеров, которые образуются при незначительном избытке антител и долгое время остаются в кровотоке. В связи с тем что СМФ и в норме фильтрует ЦИК, ее перегрузка или дисфункция увеличивают возможность персистен-ции иммунных комплексов в кровотоке и их осаждение в тканях. Кроме того, на осаждение иммунных комплексов в тканях влияют следующие факторы: заряд иммунных комплексов (анионный против катионного), валентность антигена, авидность (степень сродства антител к антигену) антитела, аффинность (сродство) антигена к компонентам различных тканей, трехмерная структура комплексов (решетка) и гемодинамические факторы.
Иммунные комплексы оседают чаще всего в почечных клубочках, суставах, коже, сердце, серозных оболочках и мелких кровеносных сосудах. Для того чтобы эти комплексы покинули систему кровообращения и осели в тканях, необходимо увеличение проницаемости сосудистого русла. Как только иммунные комплексы оседают в тканях, они инициируют острую воспалительную реакцию. В эту фазу (приблизительно через 10 дней после введения антигена) наблюдаются клинические проявления болезни (лихорадка, крапивница, артралгии, увеличение лимфатических узлов, протеинурия). Вслед за осаждением иммунных комплексов происходит активация системы комплемента с образованием ее биологически активных компонентов Активация комплемента сопровождается провоспалительными эффектами: выбросом СЗЬ-опсонина, способствующего фагоцитозу; образованием хемотаксических факторов, вызывающих миграцию полиморфно-ядерных лейкоцитов и моноцитов (С5); выбросом анафилотоксинов (СЗа и С5а), которые увеличивают проницаемость сосудов и вызывают сокращение гладких мышц, образованием комплекса (С5Ь-9), вызывающего разрушение клеточных мембран и цитолиз.
Фагоцитоз комплексов антиген — антитело лейкоцитами приводит к выбросу или образованию различных дополнительных провоспалительных веществ, включая простагландины, сосудорасширяющие белки и хемотаксические вещества. Повреждение тканей опосредуется также свободными радикалами кислорода, продуцируемыми активированными нейтрофилами. Иммунные комплексы вызывают агрегацию тромбоцитов и активацию фактора Хагемана, что приводит к усилению воспалительного процесса и образованию микротромбов. В результате развиваются васкулит, гломерулонефрит, артрит и другие болезни.
Все эти повреждения возникают при участии комплемент-связанных антител (IgG и IgM). В связи с тем что IgA может активировать комплемент по альтернативному пути, IgA-содержа-щие комплексы также могут вызывать тканевые повреждения. Важная роль комплемента в патогенезе тканевых повреждений подтверждается наблюдением, что истощение комплемента в сыворотке крови (в эксперименте) обычно уменьшает выраженность повреждений.
В морфологической картине иммунокомплексного повреждения доминирует острый некротизирующий васкулит. Например, поражение клубочков почек сопровождается гиперклеточ-ностью (большим количеством клеток) из-за набухания и пролиферации эндотелиальных и мезангиальных клеток, а также инфильтрацией нейтрофилами и моноцитами. При иммуно-флюоресцентной микроскопии иммунные комплексы видны в виде гранулярных депозитов иммуноглобулина и комплемента, а под электронным микроскопом — в виде электронно-плотных отложений (депозитов) вдоль гломерулярной базальной мембраны. Если заболевание возникает после однократного контакта с массивной дозой антигена, например при остром постстрептококковом гломерулонефрите и острой сывороточной болезни, все повреждения имеют тенденцию к разрешению благодаря катаболизму иммунных комплексов.
Хроническая сывороточная болезнь развивается при повторном или продолжительном контакте (экспозиции) с антигеном. Постоянная антигенемия необходима для развития хронической иммунокомплексной болезни, так как иммунные комплексы чаще всего оседают в сосудистом русле. Например, системная красная волчанка связана с долгим сохранением (персис-тенцией) аутоантигенов. Часто, однако, несмотря на наличие характерных морфологических изменений и других признаков, свидетельствующих о развитии иммунокомплексной болезни, антиген остается неизвестным. Такие явления характерны для ревматоидного артрита, узелкового периартериита, мембраноз-ной нефропатии и некоторых васкулитов.
2. Местная иммунокамплексная болезнь (реакция Артюса) выражается в местном некрозе ткани, возникающем вследствие острого иммунокомплексного васкулита. Этот процесс можно вызвать в эксперименте путем внутрикожного введения антигена иммунному животному, которое уже имеет циркулирующие антитела против антигена. Из-за избытка антител при попадании антигена в стенку сосудов образуются крупные иммунные комплексы, которые вызывают воспалительную реакцию. Реакция Артюса развивается в течение нескольких часов и достигает пика через 4—10 ч после инъекции, когда появляется зона видимого отека с кровоизлияниями. При иммунофлюоресцентном окрашивании удается выявить комплемент, иммуноглобулины и фибриноген, осажденные в стенках сосудов. При светооптичес-ком исследовании описывают фибриноидный некроз сосудов. Разрыв сосудов приводит к развитию местных кровоизлияний, но чаще всего наблюдается тромбоз, способствующий развитию местных ишемических повреждений.
Реакции гиперчувствительности IV типа (к л е т о ч н о-о посредованные). Такие реакции вызываются специфически сенсибилизированными Т-лимфоцитами. Они включают в себя классические замедленные реакции гиперчувствительности, вызываемые С04+Т-клетками, и прямую клеточную цитотоксичность, опосредованную С08+Т-клетка-ми. Это основной тип иммунного ответа на различные внутриклеточные микробиологические агенты, особенно микобакте-рии туберкулеза, а также на многие вирусы, грибы, простейшие и паразиты. Другими примерами являются контактная кожная чувствительность на химические вещества и реакция отторжения. Описаны два варианта реакций гиперчувствительности IV типа.
Гиперчувствительность замедленного типа (ГЗТ). Примером ГЗТ служит реакция на внутрикожно введенный туберкулин — компонент из стенок микобактерии туберкулеза. У сенсибилизированного пациента через 8—12 ч возникают покраснение и уплотнение в месте введения, а пик реакции наступает через 24—72 ч. У сильно сенсибилизированных больных в зоне инъекции развивается некроз. ГЗТ характеризуется накоплением мо-нонуклеарных клеток в подкожной ткани и дерме, преимущественно вокруг мелких вен и венул, с образованием характерных периваскулярных манжеток. Увеличение сосудистой проницаемости связано с образованием пор между эндотелиальными клетками. Выход белков плазмы за пределы сосудистого русла увеличивает отек дермы и сопровождается оседанием фибрина в интерстиции. В участках повреждения преобладают CD4+T-клетки.
При персистенции антигена макрофаги трансформируются в эпителиоидные клетки, окруженные валом из лимфоцитов, — формируется гранулема (см. главу 4). Такое воспаление характерно для IV типа гиперчувствительности и называется грануле-матозным.
Последовательность событий при ГЗТ можно рассмотреть на примере туберкулиновой реакции, которая начинается с первой встречи индивидуума с микобактериями туберкулеза. При этом CD4+-T-клетки распознают белки микобактерии туберкулеза. Происходит дифференцировка CD4+T-клеток в Thl-клетки. Дальнейшее развитие реакции зависит от цитокинов, которые секретируют Thl-клетки. Сенсибилизированные Thl-клетки поступают в кровоток и остаются там длительное время, иногда годами.
у-Интерферон является одним из наиболее важных медиаторов ГЗТ и сильным активатором макрофагов. Активированные макрофаги, обладающие способностью к фагоцитозу, уничтожают микроорганизмы. В то же время макрофаги продуцируют некоторые полипептидные факторы роста (ТцФР и ТФРр), стимулирующие пролиферацию фибробластов и усиливающие синтез ими коллагена. Таким образом, активированные макрофаги обеспечивают элиминацию антигена, а если активация продолжается, то способствуют развитию фиброза.
Цитокины ФНОа и ФНОр воздействуют на эндотелиальные клетки, вызывая повышение секреции простациклина, что приводит к увеличению кровотока в результате расширения сосудов и усиления экспрессии адгезивной молекулы Е-селектина (ELAM-1), способствующей прикреплению приходящих лимфоцитов и моноцитов. Одновременно происходит усиление секреции низкомолекулярных хемотаксических факторов, например ИЛ-8. Все эти изменения в эндотелии способствуют выходу лимфоцитов и моноцитов за пределы сосудистого русла в зону развития ГЗТ.
При цитотоксичности, опосредованной Т-лимфоцитами, сенсибилизированные С08+Т-клетки уничтожают клетки-мишени, которые являются носителями антигена (цитотоксические лимфоциты — CTL). Т-клетки, направленные против антигенов гистосовместимости, фиксированных на поверхности клеток, играют важную роль в отторжении трансплантата. Они также участвуют в защите от вирусных инфекций. В клетках, пораженных вирусом, вирусные пептиды связываются с молекулами I класса ГКГС и в виде комплексов транспортируются к поверхности клетки. Этот комплекс распознается цитотоксическими С08+Т-клетками. Лизис зараженных клеток завершается до репликации вируса, что приводит к его уничтожению. Полагают, что многие опухолевые антигены представлены на поверхности клеток, a CTL участвуют в противоопухолевом иммунитете.
3. Отторжение трансплантата. Реакция отторжения трансплантата связана с распознаванием хозяином пересаженной ткани как чужеродной. Ответственными за такое отторжение у человека являются антигены HLA. Отторжение трансплантата — сложный процесс, во время которого имеют значение как клеточный иммунитет, так и циркулирующие антитела.
Реакции, обусловленные Т-лимфоцитами. Инициация реакций, опосредованных Т-лимфоцитами, происходит тогда, когда лимфоциты реципиента встречаются с антигенами HLA донора. Полагают, что наиболее важными иммуногенами являются дендритические клетки донорских органов. Т-клетки хозяина встречаются с дендритическими клетками в пересаженном органе, а затем мигрируют в регионарные лимфатические узлы. Предшественники CD8+CTL, обладающие рецепторами к I классу HLA-антигенов, дифференцируются в зрелые CTL. В процессе дифференцировки участвуют антигенпредставляющие клетки, Т-лимфоциты и цитокины ИЛ-2, ИЛ-4 и ИЛ-5. Зрелые CTL лизируют пересаженную ткань. Кроме специфических CTL, образуются С04+Т-клетки, которые играют исключительно важную роль в отторжении трансплантата. Как и при ГЗТ, активированные С04+Т-клетки выделяют цитокины, вызывающие повышение сосудистой проницаемости и местное скопление мононуклеарных клеток (лимфоцитов и макрофагов). Считают, что ГЗТ, проявляющаяся повреждением микрососудов, ишемией и деструкцией тканей, является наиболее важным механизмом деструкции трансплантата.
Реакции, обусловленные образованием антител, могут протекать в двух вариантах. Сверхострое отторжение развивается тогда, когда в крови реципиента есть антитела против донора. Такие антитела встречаются, например, у реципиентов, у которых уже было отторжение почечного трансплантата. Предшествующие переливания крови от HLA-неидентифицированных доноров также могут привести к сенсибилизации, так как тромбоциты и лейкоциты особенно богаты HLA-антигенами. В таких случаях отторжение развивается немедленно после трансплантации, так как циркулирующие антитела образуют иммунные комплексы, оседающие в эндотелии сосудов пересаженного органа. Затем происходит фиксация комплемента и развивается реакция Артюса. У реципиентов, которые не были предварительно сенсибилизированы к антигенам трансплантата, экспозиция донорских HLA-антигенов I и II класса может вызывать образование антител. Антитела, образованные реципиентами, вызывают повреждение ткани посредством нескольких механизмов: комплементзависимой цитотоксичности, антителозави-симого, обусловленного клетками цитолиза и выпадения комплексов антиген — антитело. Изначальной мишенью для этих антител служат сосуды трансплантата. Поэтому феномен антителозависимого отторжения в почке гистологически представлен васкулитом.
Аутоиммунные болезни. Причиной некоторых заболеваний человека является развитие иммунной реакции против собственных антигенов. В норме аутоантитела могут быть найдены в сыворотке крови или тканях у многих здоровых людей, особенно в старшей возрастной группе. Безвредные антитела образуются после повреждения ткани и играют физиологическую роль в удалении ее остатков. Кроме того, нормальный иммунный ответ необходим для распознавания собственных антигенов гистосовместимости.
Различают три основных признака аутоиммунных заболеваний: 1) наличие аутоиммунной реакции, 2) наличие клинических и экспериментальных данных о том, что такая реакция не вторична к повреждению ткани, а имеет первичное патогенетическое значение, 3) отсутствие иных определенных причин болезни. Встречаются аутоиммунные болезни, при которых действие аутоантител направлено против единственного органа или ткани, и в результате развивается местное повреждение ткани. Например, при тиреоидите (зобе) Хашимото (H.Hashimoto) антитела абсолютно специфичны для щитовидной железы. При системной красной волчанке аутоантитела реагируют с составными частями ядер различных клеток. Разнообразие антител приводит к повреждениям во всем теле. При синдроме же Гуд-пасчера (E.W.Goodpasture), например, антитела против базальной мембраны легких и почек вызывают повреждения только в этих органах. Очевидно, что аутоиммунитет подразумевает потерю аутотолерантности.
Иммунологическая толерантность — это состояние, при котором иммунный ответ на специфический антиген не развивается. Состояние толерантности объясняется наличием трех механизмов: клональной делеции, клональной анергии и периферической супрессии. При клональной делеции отсутствуют саморегулирующие Ти В-лимфоциты. При клональной анергии наблюдается пролонгированная или необратимая функциональная инактивация лимфоцитов, вызванная контактом с антигеном.
Периферическая супрессия Т-лимфоцитами. Клональные де-леция и анергия являются первичными механизмами аутотолерантности (толерантности к антигенам собственного организма). Однако существуют и дополнительные механизмы. Наибольший интерес представляют супрессорные Т-лимфоциты. Супрессорные клетки могут тормозить аутореактивность, секре-тируя цитокины, снижающие интенсивность иммунного ответа.
Механизмы аутоиммунных болезней. В патогенез аутоиммунизации вовлечены иммунологические, генетические и вирусные факторы, взаимодействующие посредством сложных механизмов, которые пока мало известны.
1. Обходной путь толерантности Т-хелперов. Толерантность к аутоантигену часто связана с клональной делецией или анергией специфических Т-лимфоцитов в присутствии полностью компетентных гаптенспецифических В-лимфоцитов. Однако толерантность может быть нарушена посредством одного из следующих механизмов.
Модификация молекулы. Если потенциально слабая аутоанти-генная детерминанта (гаптен) связывается с новым носителем, то она может быть распознана нетолерантными Т-лимфоцитами как инородная. Кооперация видоизмененных гаптенов с гап-тенспецифическими В-лимфоцитами приводит к образованию аутоантител.
Молекулярная мимикрия. Некоторые инфекционные агенты перекрестно реагируют с тканями человека. Образованное в результате антитело может повреждать ткани, имеющие перекрестно реагирующие детерминанты. Поэтому ревматические заболевания сердца иногда развиваются вслед за стрептококковой инфекцией, так как антитела к стрептококковому белку М перекрестно реагируют с М-протеином в сарколемме сердечной мышцы.
Поликлональная активация лимфоцитов. В тех случаях, когда толерантность поддерживается с помощью клональной анергии, продукты их жизнедеятельности способны вызвать поликлональную (антигеннеспецифическую) активацию В-лимфоцитов. Лучше всего исследованы бактериальные липополиса-хариды (эндотоксины), которые могут индуцировать лимфоциты мышей in vitro к образованию антител против ДНК, тимоци-тов и эритроцитов.
Дисбаланс функций Т-супрессоров и Т-хелперов. Снижение функциональной активности Т-супрессоров способствует развитию аутоиммунитета и, наоборот, чрезмерная активность Т-хелперов может вызвать повышение продукции аутоантител В-лимфоцитами. Например, при системной красной волчанке наблюдается нарушение функционирования или уменьшение количества (иногда то и другое одновременно) Т-супрессоров. Активация Т-хелперов встречается также у некоторых больных системной красной Волчанкой.
Появление секвестрированного антигена. Любой аутоанти-ген, который полностью изолирован в процессе развития организма, рассматривается как инородный, если попадает в кровоток и на него развивается иммунный ответ. Сперматозоиды, основной белок миелина и кристаллин хрусталика могут попасть в категорию таких антигенов. Например, при травме яичек происходит выброс спермы в ткани, вслед за которым появляются антитела к сперматозоидам. Этот механизм аутоиммунных реакций встречается лишь в особых случаях.
Генетические факторы иммунитета. Эти факторы определяют частоту и природу аутоиммунных заболеваний. Во-первых, существует семейная предрасположенность к некоторым аутоиммунным заболеваниям человека, таким как системная красная волчанка, аутоиммунная гемолитическая анемия и аутоиммунный тиреоидит. Во-вторых, имеется связь некоторых аутоиммунных заболеваний с HLA, особенно с молекулами II класса ГКГС.
Молекулярный анализ молекул II класса ГКГС показал, что у большинства больных ревматоидным артритом имеется аллель HLA-DR4 или HLA-DR1, либо оба этих аллеля. Эти аллели имеют общий участок из 4 аминокислот, расположенный в промежутке DR-молекулы, связанном с антигеном. Таким образом, связь между ревматоидным артритом и некоторыми DR-моле-кулами может быть объяснена способностью этих последних к связыванию артритогенного антигена. Если соответствующий аллель ГКГС плохо представляет аутоантиген, то делеция соответствующего аутореактивного клона Т-лимфоцитов может и не наступить. У тех, кто наследовал такие молекулы II класса ГКГС, повышен риск развития аутоиммунитета.
Микробные агенты в аутоиммунитете. Различные микробы, микоплазмы и вирусы могут быть вовлечены в развитие аутоиммунитета. Во-первых, вирусные антигены и аутоантигены могут связываться, образуя иммуногенные единицы. Во-вторых, некоторые вирусы, например вирус Эпштейна — Барр (М.А.Ер-stein, J.M.Barr), представляют собой неспецифические, поли-клональные В-лимфоцитарные митогены и могут вызывать образование аутоантител. В-третьих, вирусная инфекция может привести к снижению функции супрессорных Т-лимфоцитов.
Вирусы и некоторые микробы, особенно бактерии (стрептококки, клебсиеллы), могут обладать эпитопами, перекрестно реагирующими с аутоантигенами. Некоторые инфекционные агенты вызывают сильную активацию и пролиферацию CD4+T-клеток. Цитокины, образованные в этих условиях, нарушают толерантность анергических Т-лимфоцитов.
Синдромы иммунного дефицита по сути представляют собой эксперименты природы, которые еще раз убеждают в сложности устройства иммунной системы. Все иммунодефици-ты делят на первичные, которые почти всегда детерминированы генетически, и вторичные, связанные с осложнениями инфекционных заболеваний, нарушенным всасыванием, старением, побочными эффектами иммуносупрессии, облучением, химиотерапией при раке и других аутоиммунных болезнях (по R.S.Cotran, V.Kumar, T.Collins, 1998).
Первичные иммунодефицит ы являются генетически детерминированными заболеваниями и поражают специфический иммунитет (гуморальный и клеточный) или неспецифические механизмы зашиты хозяина, обусловленные комплементом и клетками (фагоцитами или естественными киллерами) (схема 5.11). Несмотря на то что большинство иммуноде-фицитов встречается довольно редко, некоторые из них, например дефицит IgA, довольно-таки распространены, особенно у
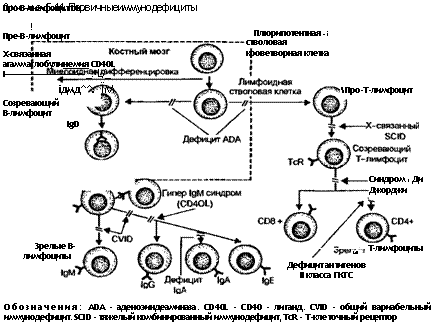 детей. Первичные иммунодефициты у детей обычно проявляются в возрасте между 6 мес и 2 годами повышенной чувствительностью к рецидивирующим инфекционным заболеваниям.
детей. Первичные иммунодефициты у детей обычно проявляются в возрасте между 6 мес и 2 годами повышенной чувствительностью к рецидивирующим инфекционным заболеваниям.
Агаммаглобулинемия Брутона (O.C.Bruton), связанная с X-хромосомой, является одним из самых распространенных первичных иммунодефицитов и характеризуется фактическим отсутствием сывороточных иммуноглобулинов, хотя наличие малых количеств IgG возможно. Это заболевание связано с X-хромосомой и встречается у лиц мужского пола. Тяжелые реци-дивируюшие инфекции обычно начинаются в возрасте 8—9 мес, когда ребенок перестает получать материнские иммуноглобулины. Чаще всего выявляются пиогенные микроорганизмы (стафилококки), больные страдают рецидивирующими конъюнктивитом, фарингитом, средним отитом, бронхитом, пневмонией и кожными инфекциями. С большинством вирусных и грибковых инфекций больной справляется успешно, так как клеточный иммунитет не нарушен. Вместе с тем существует особый риск развития процессов, связанных с вакцинацией против полиомиелита, энцефалита, вызванного вирусами ECHO, а также пневмоцистной пневмонии. Персистирующая лямблиозная инфекция приводит к нарушению всасывания.
При болезни Брутона чаще всего развиваются аутоиммунные поражения. У половины детей наблюдается патология типа ревматоидного артрита. Встречаются также системная красная волчанка, дерматомиозит и другие аутоиммунные заболевания. У этих пациентов, за редким исключением, отсутствуют В-лим-фоциты. Пре-В-лимфоциты, представляющие собой крупные лимфоидные клетки с IgM в цитоплазме, но без иммуноглобулинов на поверхности, выявляются в костном мозге в нормальных количествах. Лимфатические узлы и селезенка не имеют центров размножения. В лимфатических узлах, селезенке, костном мозге и соединительной ткани отсутствуют плазматические клетки. Небные миндалины плохо развиты или рудиментарны. В то же время имеется нормальное количество циркулирующих и тканевых Т-лимфоцитов, функция которых не изменена.
Общий вариабельный иммунодефицит представляет собой гетерогенную группу заболеваний. Он может быть врожденным или приобретенным, спорадическим или семейным (с непостоянным типом наследования). Общей особенностью всех пациентов является гипогаммаглобулинемия, обычно связанная с недостаточностью антител всех классов, но иногда только IgG. В противоположность агаммаглобулинемии Брутона в крови и лимфоидной ткани большинства больных имеется нормальное количество В-лимфоцитов. Эти В-лимфоциты, однако, не могут дифференцироваться в плазматические клетки. Чаще всего дефект выражается в терминальной дифференцировке В-лимфоцитов, в результате чего последние не могут секретировать нормальные количества иммуноглобулинов даже тогда, когда имеются хелперные Т-лимфоциты, а потенциальные супрессорные Т-лимфоциты отсутствуют.
Молекулярная основа аномальной дифференцировки В-лимфоиитов может быть разной. У некоторых больных возникают мутации, которые влияют на экспрессию иммуноглобулино-вых генов, у других — имеются дефектные В-лимфоциты, так же как и функциональные аномалии СБ4+Т-хелперов или CD8+T-cynpeccopoB. Причем количество С04+Т-клеток может быть нормальным, но они продуцируют сниженное количество ИЛ-2 и у-интерферона. В связи с тем что цитокины необходимы для секреции иммуноглобулинов, эти дефекты Т-лимфоцитов приводят к гипогаммаглобулинемии. У некоторых больных проблема может заключаться не в отсутствии Т-лимфоцитов, а скорее в абсолютном увеличении количества С08+Т-клеток, подавляющих секрецию антител нормальными В-лимфоцитами. Получены данные о генетической предрасположенности к общему вариабельному иммунодефициту.
Клинически заболевание проявляется рецидивирующими инфекциями. Гистологически наблюдается гиперплазия В-кле-точных участков лимфоидной ткани (лимфоидных фолликулов в лимфатических узлах, селезенке и кишечнике). Помимо бактериальных, эти больные страдают тяжелыми энтеровирусными инфекциями, рецидивирующим герпесом и персистирующей диареей, вызванной лямблиями. У них высока частота аутоиммунных заболеваний (около 20 \%), включая ревматоидный артрит, пернициозную и гемолитическую анемию.
Изолированный дефицит IgA очень распространен. Для заболевания характерен низкий уровень как сывороточного, так и секреторного IgA. Иммунодефицит может быть семейным или приобретенным после токсоплазмоза, кори или некоторых других вирусных инфекций. В связи с тем что IgA является основным иммуноглобулином внешней секреции, при его дефиците нарушается защита слизистых оболочек и развиваются инфекции дыхательной, желудочно-кишечной и мочеполовой систем. Больные нередко страдают синопульмональными инфекциями (сочетание синуситов и пневмоний) и диареей. У пациентов с дефицитом IgA наблюдается высокая частота аллергии респираторного тракта и различных аутоиммунных болезней, особенно системной красной волчанки и ревматоидного артрита. Причина повышенной частоты аутоиммунных и аллергических заболеваний неизвестна.
Основной причиной этого иммунодефицита является дефект дифференцировки В-лимфоцитов, продуцирующих IgA. У большинства больных с селективным дефицитом IgA имеется нормальное количество IgA-положительных В-лимфоцитов, однако большинство из них экспрессируют незрелый фенотип. Лишь немногие из этих клеток способны in vitro трансформироваться в IgA-плазматические клетки. Сывороточные антитела к IgA обнаружены приблизительно у 40 \% больных.
Синдром Ди Джорджи (A.M.DiGeorge; гипоплазия тимуса) — одна из форм селективного Т-лимфоцитарного дефицита, появление которого связано с нарушением развития третьего и четвертого глоточных карманов, дающих начало тимусу, околощитовидным железам, некоторым светлым клеткам щитовидной железы и ультимобронхиальному тельцу. Таким образом, у этих больных полностью отсутствует клеточный иммунный ответ (из-за гипоплазии или отсутствия тимуса), развиваются тетания (отсутствие околощитовидных желез) и врожденные дефекты сердца и крупных сосудов. Кроме того, внешний вид рта, ушей и лица могут быть ненормальными. Отсутствие клеточного иммунитета отражается в низком уровне циркулирующих Т-лим-фоцитов и слабой защите против некоторых грибковых и вирусных инфекций. Плазматические клетки в лимфоидной ткани представлены в нормальных количествах, но тимусзависимые паракортикальные зоны лимфатических узлов и периартерио-лярных оболочек в селезенке отсутствуют. Уровни иммуноглобулинов в пределах нормы.
Синдром Ди Джорджи не относится к числу генетически детерминированных заболеваний, но, по-видимому, является результатом внутриматочного повреждения плода на 8-й неделе беременности.
Тяжелые комбинированные иммунодефицитные заболевания характеризуются комбинированным Ви Т-лимфоцитарным дефектом. Больные дети страдают от тяжелых рецидивирующих инфекций. Среди возбудителей следует назвать Candida albicans, Pneumocystis carinii, Pseudomonas, а также цитомегаловирус, вирус ветряной оспы и многие другие бактерии. Без пересадки костного мозга смерть наступает в первые годы жизни.
В зависимости от локализации мутантного гена и природы генетического дефекта различают два типа наследования — ау-тосомно-рецессивный и рецессивный, связанный с Х-хромосо-мой. Среди аутосомно-рецессивных форм заболевания приблизительно у 40 \% больных отсутствует фермент аденозиндеами-наза, дефицит которого ведет к накоплению диоксиаминазина и его производных, особенно токсичных для незрелых лимфоцитов, в первую очередь Т-лимфоцитов. Реже при аутосомно-ре-цессивном типе этого заболевания встречается дефект активации Т-лимфоцитов. У этих больных имеется нормальное количество Т-лимфоцитов, однако существует дефицит одной из нескольких молекул, участвующих в активации этих клеток. Приблизительно у 50 \% больных встречается рецессивный тип наследования, связанный с Х-хромосомой. У этих больных происходит мутация, которая воздействует на белок, являющийся рецептором для ИЛ-2, ИЛ-4 и ИЛ-7.
Характер морфологических изменений зависит от вида генетического дефекта. При двух наиболее распространенных формах иммунодефицита (отсутствие аденозиндиаминазы и мутация рецепторов) тимус малых размеров, лишен лимфоидных клеток. В других случаях лимфоидная ткань гипопластична с заметным уменьшением размеров зон Т-лимфоцитов, а в некоторых случаях как Т-, так и В-зон.
Иммунодефицит с тромбоцитопенией и экземой [синдром Вискотта—Олдрича (A.Wiskott, R.A.Aldrich)] — рецессивное, связанное с Х-хромосомой заболевание, которое характеризуется тромбоцитопенией, экземой, восприимчивостью к рецидивирующей инфекции и рано заканчивается смертью. Тимус морфологически нормальный, однако наблюдается прогрессирующее вторичное истощение Т-лимфоцитов в периферической крови и паракортикальных (тимусзависимых) зонах лимфатических узлов с одновременным снижением клеточного иммунитета. Уровень IgM в сыворотке крови низкий, однако уровень IgG обычно нормальный. У больных часто развиваются злокачественные лимфомы.
Генетический дефицит системы комплемента описан для всех компонентов системы комплемента и двух ее ингибиторов. Дефицит компонентов комплемента, особенно СЗ, который необходим как для классического, так и альтернативного пути его активации, вызывает повышенную чувствительность к инфекции патогенными бактериями. Врожденный дефицит Clq, С2 и
С4 повышает риск развития иммунокомплексных заболеваний, например системной красной волчанки. Отсутствие ингибитора С1-эстеразы вызывает неконтролируемую активацию С1-эсте-разы с образованием кинина С2. У этих больных развивается врожденный ангионевротический отек, характеризующийся местным отеком пораженной кожи и слизистых оболочек. Дефицит компонентов классического пути (С5—8) вызывает рецидивирующие нейссериальные (гонококковые, менингококко-вые) инфекции.
8. Синдром приобретенного иммунодефицита (СПИД). Эпидемиология. К началу XXI века СПИД зарегистрирован в более чем 165 странах мира, а наибольшее количество инфицированных лиц находится в Африке и Азии. Идентифицированы 5 групп риска среди взрослых людей: гомосексуальные и бисексуальные мужчины составляют наиболее крупную группу (до 60 \% больных); лица, которые вводят внутривенно наркотики (до 23 \%); больные гемофилией (до 1 \%); реципиенты крови и ее компонентов (около 2 \%); гетеросексуальные контакты членов других групп повышенного риска, главным образом наркоманов (6 \%). Приблизительно в 6 \% случаев факторы риска не определяются. Около 2 \% больных СПИДом дети. В 80 \% случаев заражение детей происходит от матери, в 20 \% — отмечается гемофилия.
Этиология. Возбудителем СП ИДа является вирус иммунодефицита человека — ретровирус, относящийся к семейству лентивирусов, включающему также вирусы кошачьего иммунодефицита, висны овец, конской анемии. Эти ретровирусы имеют некоторые общие особенности: длительный инкубационный период, тропизм к кроветворной и нервной системе, вызывают иммуносупрессию, цитопатические эффекты in vitro. Различают две генетически разные формы вируса СПИДа — вирусы иммунодефицита человека 1 и 2 (HIV-1 и HIV-2). HIV-1 — наиболее распространенный тип, встречается в США, Европе и Центральной Африке, a HIV-2 — главным образом в Западной Африке.
Патогенез. Существуют две основные мишени для вируса СПИДа: иммунная система и ЦНС. Иммунопатогенез СПИДа характеризуется развитием глубокой иммунодепрессии, что главным образом связано с выраженным уменьшением количества С04+Т-клеток. Имеется множество доказательств того, что молекула CD4 фактически является высокоаффинным рецептором для вируса СПИДа. Это объясняет селективный тропизм вируса к С04+Т-клеткам. Инфекция начинается со связывания гликопротеина оболочки вируса gpl20 с молекулами CD4. Затем происходят слияние вируса с клеточной мембраной и его интернализация. После интернализации вируса геном клетки подвергается обратной транскрипции, что приводит к образованию провирусной ДНК.
8. Пальцев, т. 1.
225
В покоящихся Т-лимфоцитах вирус и провирусная ДНК могут оставаться в цитоплазме в линейной эписомальной форме. В делящихся Т-лимфоцитах провирусная ДНК входит в ядро, а затем интегрируется в геном хозяина. После этого про-вирус может оставаться в хромосоме в течение нескольких месяцев и лет, и с этих пор инфекция может стать латентной. Провирусная ДНК, наоборот, может быть транскрибирована с образованием полноценных вирусных частиц, которые отпочковываются от клеточной мембраны. Инфекция, связанная с интенсивным отпочковыванием вируса, ведет к смерти клетки. Инициация транскрипции провирусной ДНК происходит под влиянием антигенов или цитокинов.
Кроме гибели инфицированных С04+Т-клеток, существуют и другие непрямые механизмы развития заболевания. Во-первых, происходит уменьшение количества незрелых предшественников С04+Т-клеток, связанное с их прямым инфицированием в тимусе, а также с инфицированием клеток, секретирую-щих цитокины, необходимые для дифференцировки CD4+T-клеток. Во-вторых, наблюдается слияние инфицированных и неинфицированных клеток с образованием синцития (гигантских клеток), происходит аутоиммунная деструкция как инфицированных, так и неинфицированных С04+Т-клеток. В связи с тем что многие больные имеют циркулирующие антитела к gpl20, клетки с наличием на поверхности gpl20 могут быть разрушены. Уменьшение количества С04+Т-клеток вызывает изменение соотношения CD4—CD8 в периферической крови. Возникают также качественные дефекты Т-клеток. Например, наблюдаются снижение пролиферации Т-клеток, вызванной антигеном, падение выработки цитокинов ИЛ-2 и у-интерферо-на, дефекты во внутриклеточной передаче.
Кроме того, инфицирование моноцитов и макрофагов является исключительно важным для патогенеза СПИДа. Как и Т-лимфоциты, большинство макрофагов, инфицированных вирусом иммунодефицита, образованы в тканях, а не в периферической крови. Многие макрофаги экспрессируют малые количества CD4, а вирус может инфицировать эти клетки посредством gpl20-CD4-MexaHH3Ma. Кроме того, вирус может проникать в макрофаги с помощью фагоцитоза или эндоцитоза его частиц, покрытых антителами, с помощью Fc-рецепторов. Инфицированные макрофаги отпочковывают относительно малые количества вируса, но эти клетки содержат большие количества вирусных частиц, расположенных исключительно во внутриклеточных вакуолях. Несмотря на то что в макрофагах возможна репликация вируса, в отличие от С04+Т-клеток они совершенно резистентны к цитоплазматическому действию вируса.
Инфицирование макрофагов приводит к тому, что моноциты и макрофаги превращаются в настоящую фабрику по производству вирусов и резервуар для их хранения. Кроме того, мак-
рофаги способны транспортировать вирус по всему телу, особенно в нервную систему. В отличие от тканевых макрофагов количество моноцитов в кровотоке снижается. Одновременно снижаются противомикробная активность, хемотаксис, секреция ИЛ-1, ФНОа, способность представлять антигены Т-лимфоцитам. Важным резервуаром вируса являются также дендритические клетки в центрах размножения лимфатических узлов.
Таким образом, СБ4+Т-клетки, макрофаги и дендритические клетки, а не клетки крови, являются главными резервуарами вируса. Кроме того, у больных СПИДом развиваются глубокие нарушения функционирования В-лимфоцитов. Так, у этих больных наблюдаются гипергаммаглобулинемия и циркулирующие иммунные комплексы (ЦИК), связанные с поликлональ-ной активацией В-лимфоцитов. Среди причин этого феномена называют инфицирование В-лимфоцитов цитомегаловирусом и вирусом Эпстайна — Барр, каждый из которых является поли-клональным активатором В-лимфоцитов. Gpl20 сам по себе может вызывать рост и дифференцировку В-лимфоцитов, инфицированные вирусом СПИДа макрофаги образуют повышенные количества ИЛ-6, которые способствуют активации В-лимфоцитов. Несмотря на наличие спонтанно активированных В-лимфоцитов, больные СПИДом не способны поддерживать антительный ответ на новый антиген. Поэтому больные СПИДом подвержены диссеминированным инфекциям, вызванным инкапсулированными бактериями.
Макрофаги и микроглия (клетки, относящиеся к моноцитам и макрофагам) являются основными типами клеток мозга, которые инфицируются вирусом иммунодефицита человека. Полагают, что инфицированные макрофаги продуцируют растворимые факторы, которые могут быть токсичными для нейронов или нарушать их функции без прямой токсичности. Среди таких растворимых факторов выделяют цитокины (ИЛ-1). Известно также о прямом повреждении нейронов посредством растворимого gpi20.
Сравнительное изучение возбудителя, выделенного из мозга, с вирусом из лимфоцитов показывает, что первый из них содержит специальные подгруппы вируса СПИДа. Эти различия в тропизме вируса связывают со специфическим доменом молекулы gpl20.
Течение синдрома приобретенного иммунодефицита (СПИД) складывается из трех фаз, отражающих динамику взаимодействия вируса с хозяином: ранней острой фазы, средней хронической и финальной кризисной фаз. В раннюю фазу развивается первоначальный ответ иммуно-компетентного индивидуума на вирус. Она характеризуется высоким уровнем образования вируса, виремией и распространенным обсеменением лимфоидной ткани. В этот период, однако, инфекция контролируется с помощью антивирусного иммунного ответа. Хроническая фаза представляет собой период относи-
8*
227
тельного сдерживания вируса, когда иммунная система интакт-на, но наблюдается слабая репликация вируса, преимущественно в лимфоидной ткани. Эта фаза может продолжаться несколько лет. Финальная фаза характеризуется нарушением защитных механизмов хозяина и безудержной репликацией вируса. Снижается содержание С04+Т-клеток. После неустойчивого периода появляются серьезные оппортунистические инфекции, вторичные опухоли, признаки неврологического заболевания.
Амилоидоз. Амилоид представляет собой белок, который откладывается между клетками в различных тканях и органах. Его распознавание в клинике зависит исключительно от обнаружения в биоптатах. При светооптическом исследовании с использованием традиционных окрасок амилоид выглядит как аморфное, эозинофильное, гиалиноподобное межклеточное вещество, в результате прогрессирующего накопления и давления которого развивается атрофия клеток. Для того чтобы отличить амилоид от других депозитов (коллаген, фибрин), используют ряд гистохимических методов, например окраску конго красным. В поляризационном микроскопе амилоид зеленоватого цвета и дает двойное лучепреломление.
Несмотря на то что все депозиты имеют одинаковый вид и тинкториальные свойства, химически амилоид неоднороден. Различают две основные и несколько малых биохимических форм. Они образуются с участием разных патогенетических механизмов. Видимо, амилоидоз представляет собой группу заболеваний, основным признаком которой является отложение похожих веществ белкового строения.
Физическая природа амилоида. При электронной микроскопии амилоид состоит из неветвящихся фибрилл длиной приблизительно 7,5—10 нм. Эта структура амилоида одинакова при всех видах амилоидоза. При кристаллографии и инфракрасной спектроскопии обнаружено характерное складчатое строение оболочки. Эта особенность строения и объясняет появление двойного лучепреломления. Кроме того, в меньших количествах выявлен и второй компонент (Р-компонент), который имеет пентагональное строение.
Химическая природа амилоида. Приблизительно 95 \% амилоида состоит из фибриллярного белка, остальные 5 \% остаются на долю гликопротеинового Р-компонента. Среди 15 различных биохимических вариантов амилоидного белка выделены два основных: амилоид из легких цепей (AL), который образуется плазматическими клетками (иммуноцита-ми) и содержит легкие цепи иммуноглобулина; связанный амилоид (АА) — уникальный неиммуноглобулиновый белок, синтезированный печенью. Эти два амилоидных белка возникают в разных условиях. AL-амилоид образуется из МН2-терминальных фрагментов легких цепей иммуноглобулинов. Большая часть AL-амилоида состоит из Я.-легких цепей (особенно VI типа) или их фрагментов, однако в некоторых случаях идентифицированы к-цепи. AL-амилоид образуется иммунноглобулинсекретирую-щими клетками при некоторых формах моноклональной пролиферации В-клеток.
Молекула АА-амилоида имеет мол. массу 8500 и состоит из 76 аминокислотных остатков. АА-фибриллы образуются из более крупных предшественников, циркулирующих в крови (амилоид, связанный с сывороткой крови) — SAA. Они синтезируются в печени и циркулируют вместе с HDL3 — субклассами липопротеинов. АА-белок образуется при вторичном амило-идозе.
В депозитах амилоида обнаруживают и другие белки. Транс-тиретин — нормальный белок сыворотки, который связывает и транспортирует тироксин и ретинол. Мутантная форма трансти-ретина (и его фрагменты) обнаруживаются при генетически детерминированных заболеваниях, которые называют семейной амилоидной полинейропатией. Амилоидный транстиретин (ATTR) отличается от нормального единственной аминокислотой. р2-Амилоид — пептид, составляющий ядро мозговых бляшек при болезни Альцгеймера; он образуется из наиболее крупных трансмембранных гликопротеидов, так называемых белковых предшественников амилоида (АРР). Встречаются также депозиты амилоида, образованные из разных предшественников, таких как гормоны (прокальцитонин) и кератин.
Р-компонент отличается от амилоидных фибрилл, но при всех формах амилоидоза тесно с ними связан. Он обладает структурной гомологией с С-реактивным белком. Сывороточный Р-компонент обладает сродством к фибриллам амилоида и необходим для образования депозитов в тканях.
Классификация амилоидоза основана на химическом строении амилоида (AL, АА, ATTR) и клинических синдромах. Амилоидоз может быть системным (генерализованным) с поражением нескольких систем органов или местным, когда депозиты обнаруживаются только в одном органе.
Системный (генерализованный) амилоидоз бывает первичным, если связан с дискразией иммуноцитов, или вторичным, когда возникает как осложнение хронического воспаления или деструктивных процессов в тканях. Врожденный (семейный) амилоидоз образует отдельную гетерогенную группу.
Дискразия иммуноцитов с амилоидозом (первичный амилоидоз). Этот тип амилоидоза обычно носит системный характер, состоит из AL-амилоида и составляет около 75 \% всех наблюдений этого заболевания. В основе его лежит развитие дискразии плазматических клеток. Он встречается у больных с множественной миеломой, для которой характерны множественные остеолити-ческие повреждения скелета. Необходимым, хотя и недостаточным, условием развития амилоидоза является наличие белка Бене-Джонса, обладающего только легкими цепями.
Реактивный системный амилоидоз. Для этого вида амилоидо-за характерно образование АА-амилоида. Его также называют вторичным амилоидозом, так как он связан с хроническим воспалением, сопровождающимся разрушением тканей. Амилоидоз встречается при туберкулезе, бронхоэктазии, хроническом остеомиелите. Однако при эффективной антимикробной терапии условия для развития амилоидоза могут исчезнуть. Чаще всего реактивный системный амилоидоз осложняет течение ревматоидного артрита и других заболеваний соединительной ткани, таких как анкилозирующий спондилит, воспалительные заболевания кишечника, особенно региональный энтерит и язвенный колит. Амилоидоз развивается приблизительно у 3 \% больных ревматоидным артритом.
Амилоидоз, связанный с гемодиализом. Возникает у больных после длительного гемодиализа, проводимого в связи с почечной недостаточностью, и вследствие выпадения р2-микроглобу-лина. Этот белок выявляется в больших количествах в сыворотке крови нефрологических больных, так как не фильтруется через диализные мембраны. Примерно у 70 \% больных имеются депозиты амилоида в синовии, суставах и сухожилиях.
Врожденный семейный амилоидоз. Это относительно редкое заболевание и встречается в определенных географических районах. Лучше всего исследован аутосомно-рецессивный вариант семейного амилоидоза, который называют семейной средиземноморской лихорадкой. Клинически это заболевание характеризуется приступами лихорадки, сопровождающимися воспалением серозных оболочек, включая брюшину, плевру и синовиальные оболочки. Встречается обычно у армян, сефардов и арабов. Амилоид при этом заболевании представлен АА-вариантом.
В отличие от аутосомно-рецессивного варианта аутосомно-доминантный семейный амилоидоз характеризуется выпадением амилоида преимущественно в периферических нервах. Семейная амилоидная полинейропатия описана в разных регионах мира. Например, невропатический амилоидоз обнаружен в Португалии, Японии, Швеции и США. При всех этих генетических заболеваниях фибриллы амилоида состоят из ATTR.
Локализованный амилоидоз. Депозиты амилоида обычно проявляются в виде узелков, видимых только под микроскопом, как правило, только в одном органе. Опухолеподобные депозиты амилоида чаще всего встречаются в легких, гортани, коже, мочевом пузыре, языке и около глаз. Часто на периферии амилоидных масс находят инфильтрацию лимфоцитами и плазматическими клетками, которую расценивают как ответ на выпадение амилоида. В некоторых случаях амилоид состоит из AL-белка и имеет иммуноцитарное происхождение.
Эндокринный амилоидоз. Микроскопические депозиты амилоида иногда обнаруживаются в некоторых эндокринных опухолях, таких как медуллярная карцинома, опухоли островков под-
желудочной железы, феохромоцитома, низкодифференцирован-ные карциномы желудка, а также в островках Лангерганса поджелудочной железы при сахарном диабете II типа. В этих случаях амилоидогенные белки образуются из полипептидных гормонов, например амилоидный полипептид островков (IAPP) поджелудочной железы.
Амилоид старения. При старении встречаются два вида амилоидных депозитов. Старческий сердечный амилоидоз характеризуется выпадением в сердце у престарелых больных (обычно в возрасте 80—90 лет). Он встречается в двух формах: выпадение транстиретина, вовлекающего желудочки, или выпадение атри-ального натрийуретического пептида, повреждающего предсердие. Заболевание развивается часто бессимптомно, но может вызывать тяжелые нарушения сердечной деятельности. Одновременно обнаруживаются депозиты амилоида в легких, поджелудочной железе, селезенке. Это позволяет предполагать, что старческий амилоидоз является системным заболеванием.
Старческий церебральный амилоидоз развивается в результате отложений депозитов АВ2-белка в мозговые кровеносные сосуды и бляшки у больных болезнью Альцгеймера.
Патогенез. Несмотря на то что предшественники двух основных амилоидных белков идентифицированы, некоторые аспекты их происхождения еще не ясны. При реактивном системном амилоидозе имеет значение длительное разрушение тканей и воспаление, которые приводят к повышению уровня SAA в сыворотке крови. SAA синтезируется клетками печени под влиянием цитокинов ИЛ-1 и ИЛ-6. Повышенный уровень SAA характерен для воспаления, но в большинстве случаев не приводит к амилоидозу. SAA в норме разрушается до конечных растворимых продуктов под действием ферментов моноцитов. Больные, у которых развивается амилоидоз, видимо, имеют дефект этого фермента, что приводит к неполному разрушению SAA и формированию нерастворимой молекулы АА. Наоборот, генетически детерминированные структурные аномалии в молекуле SAA сами по себе вызывают ее резистентность к разрушению моноцитами. В случае иммуноцитарной дискразии обнаружен излишек легких цепей иммуноглобулинов, а амилоид может образовываться в результате протеолиза легких цепей иммуноглобулинов. Неполноценная деградация приводит к образованию легких цепей, резистентных к полному протеолизу.
При семейном амилоидозе выпадение транстиретина в виде амилоидных фибрилл не является следствием гиперпродукции транстиретинов. Полагают, что генетически детерминированные повреждения структуры подталкивают к образованию транстиретинов, склонных к аномальной агрегации и протеолизу.
Клетки, участвующие в превращении белков-предшественников в фибриллы, не полностью охарактеризованы, но «основными кандидатами» на выполнение этих функций являются макрофаги.
Глава 6.

Обсуждение Патологическая анатомия
Комментарии, рецензии и отзывы