8.5. цитогенетические заболевания
8.5. цитогенетические заболевания
Аберрации хромосом, лежащие в основе цитогенетических заболеваний, — хромосомные мутации — могут выражаться в изменении числа или нарушении структуры хромосом.
Обычными причинами анэуплоидии служит неразъединение хромосом при делении либо задержка удвоения на уровне анафазы. Первая из этих причин действует, когда гомологичная пара хромосом не расходится во время первого деления при мейозе или же две хроматиды не расходятся во втором мейоти-ческом делении либо при делениях соматических клеток. Все это приводит к формированию двух анэуплоидных клеток. Когда в ходе гаметогенеза происходит такое нерасхождение, образующиеся гаметы имеют в своем хромосомном наборе одну лишнюю хромосому (п + 1) или теряет одну хромосому (п — 1). Оплодотворение таких клеток нормальными гаметами может приводить к образованию двух типов зигот: с трисомией (2п +1) или моносомией (2п — 1).
При анафазной задержке в мейозе «отстает» одна гомологичная хромосома или в митозе — одна хроматида. Затем они покидают ядро клетки. В результате появляются одна нормальная клетка и одна с моносомией. Моносомия и трисомия половых хромосом, а также более причудливые аберрации совместимы с жизнью и обычно связаны с различными уровнями нарушений фенотипа. Моносомия, возникающая у лиц с аутосомией, выражается в утрате избыточной генетической информации, что позволяет осуществиться эмбриогенезу и живорождению, а несколько аутосомных трисомий дают возможность лишь для выживания. Кроме трисомий 21, все прочие приводят к тяжелым дефектам у детей, которые почти все без исключения погибают на 1-м году жизни.
Иногда ошибки, происходящие во время митоза на ранних стадиях развития какого-либо организма, дают начало двум или более популяциям развивающихся клеток. Это заболевание называется мозаичностъю, или мозащизмом. Оно может развиваться после ошибок в митозе, во время дробления оплодотворенной яйцеклетки либо в соматических клетках.
Мозаичность, поражающая половые хромосомы, встречается довольно часто. При делении оплодотворенной яйцеклетки ошибка приводит к формированию дочерней клетки с тремя половыми хромосомами, в то время как другая клетка получает только одну такую хромосому. Создается мозаичность по хромосомному набору (или числу): 45,Х/47,ХХХ. Все клетки — потомки таких предшественников имеют набор либо 47,ХХХ, либо 45,Х. Патология у человека с таким числом хромосом может представлять собой вариант синдрома Тернера (H.H.Turner; — отсутствие у женщины одной Х-хромосомы в кариотипе). При этом выраженность фенотипической экспрессии зависит от количества и распределения в организме клеток с набором 45,Х. Если ошибка случается при более позднем дроблении, то мозаичность проявится благодаря трем клеточным популяциям с наборами 45,Х / 46,ХХ / 47,ХХХ. Один из этих наборов — 45,ХХ — будет нормальным. Повторные ошибки в митозе увеличивают количество популяций с разным кариотипом.
Аутосомная мозаичность встречается значительно реже, чем аберрации половых хромосом. Ошибка в ходе раннего митоти-ческого деления, поражающая аутосомы, обычно приводит к мозаичности, не совместимой с жизнью и включающей в себя аутосомную моносомию. Изредка в период эмбриогенеза организм переносит утрату нежизнеспособной клетки и возникновение мозаичности 46,XY/47,XY+21. У такого ребенка появляется мозаичность трисомий 21с частично выраженным синдромом Дауна (степень выраженности зависит от количества клеток с трисомией).
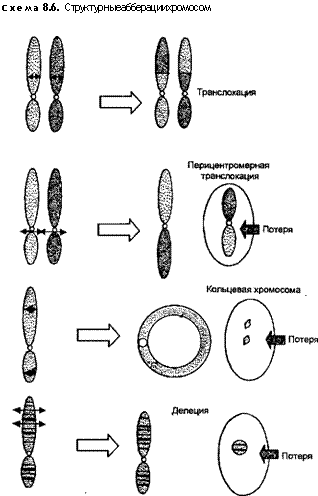
Для обнаружения структурных аберраций хромосом с помощью современной техники бендинга (о бендинге см. главу 1 и раздел 8.1), нужно, чтобы эти аберрации имели значительный объем и включали в себя большой участок ДНК (около 4 млн пар оснований), содержащий несколько генов. Структурные изменения, как правило, являются следствием полома хромосом, сопровождающегося утратой или перераспределением генетического материала (схема 8.6). В небольшом количестве эти из
 |
менения могут происходить спонтанно, но иногда их частота возрастает под влиянием мутагенов окружающей среды, главным образом всевозможных химических соединений, а также ионизирующей радиации (по R.S.Cotran, V.Kumar, T.Collins, 1988). Кроме того, несколько редких аутосомно-рецессивных генетических заболеваний (анемия Фанкони, синдром Блума и атаксия-телеангиэктазия) связаны с таким высоким уровнем нестабильности структуры хромосом, что их называют синдромами полома хромосом.
Утрата какой-либо части хромосомы называется делецией. Она может быть терминальной или интерстициальной. Терминальные делеции происходят в результате одиночного полома в каком-то плече, при этом возникает фрагмент без центромеры, который при очередном делении клетки исчезает. Так, при утрате части короткого плеча хромосомы 16 это изменение полностью обозначается как 46,XY,16p-. Часто нужно указать поточнее, в каком районе и в какой полосе произошла делеция. Пример — 46,XY, del(16)(p) — указывает, что точка полома локализуется в зоне полосы 4, района 1 короткого плеча хромосомы 16. Интерстициальные делеции происходят в том случае, когда исчезает участок какого-либо плеча хромосомы, располагающийся между двумя точками полома.
Еще один вид аберраций — транслокация — выражается в переносе сегмента одной хромосомы на любую другую. При одной ее разновидности, названной уравновешенной реципрок-ной транслокацией, в каждой из двух разных хромосом имеется по одиночному полюсу и происходит взаимообмен генетическим материалом. Эта аберрация может быть нераспознана без применения бендинга. Уравновешенная реципрокная транслокация между длинным плечом хромосомы 2 и коротким плечом хромосомы 5 обозначается как 46,XX,t(2;5)(q31;pl4). Расшифровка: у данного человека 46 хромосом; у одной из хромосом пары 2 и у одной — пары 5 строение изменено. Поскольку утрата генетического материала не произошла, этот человек остается фенотипически нормальным. Однако у данного носителя уравновешенной транслокации имеется повышенный риск воспроизведения ненормальных гамет. В приведенном случае может образоваться гамета, содержащая одну нормальную хромосому 2 и одну хромосому 5 с транслокацией. Такая гамета не была бы генетически уравновешенной, так как не содержала бы нормальный набор генетического материала. Последующее оплодотворение нормальной гаметой привело бы к формированию ненормальной (неуравновешенной) зиготы, что выразилось бы в спонтанном аборте или рождении больного ребенка.
Еще одна важная разновидность транслокации, названная робертсоновской (J.Robertson), или слиянием центромер, представляет собой реципрокную транслокацию между двумя акро-центрическими хромосомами. Обычно разрыв происходит вблизи центромер каждой хромосомы, затрагивая длинное плечо у одной и короткое — у другой. Следующий за разрывом перенос генетического материала влечет за собой появление одной очень большой и одной совсем мелкой хромосом. Последняя нередко исчезает, и коль скоро она несет так мало генетической информации, эта утрата является совместимой с нормальным фенотипом. Поэтому робертсоновская транслокация между двумя хромосомами обнаруживается неожиданно и у явно здоровых лиц. Значение же этого повреждения заключается в воспроизводстве ненормального потомства.
Следующим видом аберраций является формирование изо-хромосомы. При этом одно плечо хромосомы утрачивается, а оставшееся удваивается, что приводит к образованию хромосомы либо с двумя короткими, либо с двумя длинными плечами. Изо-хромосома содержит генетическую информацию, морфологически идентичную в обоих плечах. Наиболее частый вариант изохромосомы, встречающийся у живорожденных, выражается в изменении длинного плеча хромосомы X и обозначается как i(Xq). Этот вариант связан с моносомией генов короткого плеча хромосомы X и трисомией генов длинного ее плеча.
Кольцевая хромосома — особая форма делеции — образуется, когда делеция встречается на обоих концах хромосомы и происходит слияние поврежденных концов. Если при этом утрачен значительный генетический материал, то развиваются нарушения фенотипа. Пример: 46,XY,r(14). Кольцевые хромосомы не проходят нормально через мейоз или митоз, что обычно приводит к серьезным последствиям.
Наконец, еще один вид аберраций — инверсия — в перераспределении генетического материала. Это происходит в результате разрыва двух участков в одной хромосоме с последующей реин-корпорацией (включением снова) сегмента, перевернутого на 180°. Инверсия, происходящая только в одном плече, называется парацентрической. Если же разрывы возникают на противоположных от центромеры плечах, то инверсия становится перицент-рической. Инверсии практически не влияют на нормальное развитие.
В специальной литературе описано значительно больше количественных и структурных аберраций, число открываемых ненормальных кариотипов, встречающихся при генетических болезнях, возрастает с каждым годом. Но все же клиническое значение хромосомных нарушений находится на стадии начального изучения.По современным данным, около 7,5 \% всех зачатий сопровождается хромосомными аберрациями, большинство из которых несовместимо с живорождением или выживанием. Так, указанные нарушения выявлены при 50 \% спонтанных абортов, у 5 \% мертворожденных и новорожденных, умирающих непосредственно после рождения. Даже у живорожденных частота аберраций колеблется между 9,5 и 1,0 \%.
Онтогенетические заболевания, связанные с аутосомами. Трисомия 21 (синдром Дауна; J.Down). Синдром Дауна — наиболее частое хромосомное заболевание человека и основная причина умственной отсталости. В США распространенность его среди новорожденных достигает 1:800. Примерно 95 \% пораженных индивидуумов имеют трисомию 21 (рис. 8.5), таким образом, их хромосомное число равно 47. Большинство других имеют нормальное хромосомное число, но также излишний хромосомный материал, появившийся в результате транслокации. Как уже упоминалось, самой частой причиной трисомии и,

следовательно, синдрома Дауна, является нерасхождение хромосом в ходе мейоза. У родителей таких детей кариотип нормален, и сами они могут быть полностью нормальны.
Возраст матери существенно влияет на частоту синдрома Дауна. По статистическим данным за 1991 г., этот синдром встречается у 1 новорожденного из 1550 живорожденных у матерей младше 20 лет и соответственное у 1 из 25 у матерей старше 45 лет. Такое соотношение заставляет предполагать, что в большинстве случаев в яйцеклетке во время мейоза происходит нерасхождение хромосомы 21. Исследования с использованием полиморфной ДНК для метки родительской хромосомы 21 показали, что в 95 \% случаев трисомии лишняя хромосома 21 имеет материнское происхождение. Несмотря на множество высказанных гипотез, причина возросшей восприимчивости яйцеклетки к указанному нерасхождению неизвестна.
Примерно у 4 \% больных синдромом Дауна лишний хромосомный материал образуется после робертсоновской транслокации длинного плеча хромосомы 21 на другую акроцентрическую хромосому, например 22 или 14. Поскольку оплодотворенная яйцеклетка уже обладает двумя нормальными аутосомами 21, материал, перемещенный при транслокации, обеспечивает ту же тройную «дозу» генов, как при трисомий 21. Подобные случаи часто (но не всегда) имеют семейный характер, и перемещенная хромосома наследуется от одного из родителей, являющегося носителем робертсоновской транслокации. В качестве примера можно привести кариотип с материнским носительст-вом: 45,XX,-14,-21,+t(14q 21q). В тех случаях, когда ни один родитель не является носителем заболевания, перераспределение генетического материала происходит в ходе гаметогенеза. Теоретически родитель-носитель имеет 1 шанс из 3 «получить» живорожденного ребенка с синдромом Дауна, однако частота поражения детей в таких ситуациях гораздо ниже. И причины этого расхождения пока непонятны.
Около 1 \% больных синдромом Дауна имеют мозаичность, которая обычно проявляется у них в смешанной клеточной популяции с 46 и 47 хромосомами. Мозаичность формируется в результате митотического нерасхождения хромосомы 21 на ранней стадии эмбриогенеза. У таких больных симптоматика разнообразна и более умеренна (по сравнению с другими пациентами, имеющими тот же синдром), что зависит от числа ненормальных клеток. Несомненно, в случаях транслокации или мозаичности при синдроме Дауна возраст матери не имеет значения.
Клинико-диагностические признаки синдрома совершенно очевидны уже при рождении: уплощенное лицо в профиль, косой разрез глаз, вертикальная кожная складка, прикрывающая медиальный угол глазной щели (эпикантус). Такие признаки отражают устаревшие и неудачные названия «монголизм» или «монголоидная идиотия». Среди причин слабоумия синдром Дауна — ведущая. Развивается тяжелая умственная отсталость: примерно 80 \% пораженных имеют показатель умственного развития (IQ — intelligence quotient) в пределах 25—50. Но ирония судьбы состоит в том, что такие дети имеют мягкий характер, застенчивы и управляемы значительно легче, нежели их более удачливые братья и сестры, менее отягощенные хромосомными нарушениями. Следует отметить, что у некоторых носителей мозаичности и синдрома Дауна фенотип изменен незначительно, у них может быть нормальное или почти нормальное умственное развитие.
Упомянем о некоторых других клинических признаках заболевания. Около 40 \% больных имеют врожденные поражения сердца, чаще всего дефекты эмбриональной закладки эндокарда, включающие дефект нижней части межпредсердной перегородки (ostium primum) или другой ее части [кроме зоны овальной ямки (дефект называется ostium secundum) и верхней зоны (sinus venosus)], пороки атриовентрикулярных клапанов и дефекты межжелудочковой перегородки. Большинство смертельных исходов в периоде новорожденное™ и раннем детстве обусловлены именно этими дефектами.
У детей с трисомией 21 в 10—20 раз повышен риск развития острого лейкоза. У них наблюдаются и острый лимфобластный, и острый нелимфобластный варианты. Относительная частота каждого из этих вариантов сходна у детей с синдромом Дауна и у детей без такого синдрома. Фактически у всех больных с трисомией 21, «перешагнувших» 40-летний возраст, развиваются неврологические изменения, характерные для болезни Альцгей-мера, формы сенильной деменции (старческого слабоумия). Кроме того, отмечаются нарушения иммунной реактивности, предрасполагающие к появлению серьезных инфекций (в частности в легких), а также к развитию аутоиммунного тироидита. Несмотря на то что описано несколько нарушений, возникающих в основном в субпопуляциях Т-лимфоцитов, клеточные основы иммунологических расстройств остаются неясными. И все же более 80 \% больных с трисомией 21 живут при налаженных медицинской помощи и уходе до 30 лет и более.
И кариотип, и симптоматика трисомии 21 хорошо известны в течение более 30 лет. Но о молекулярных механизмах синдрома Дауна сведений мало. При исследовании у больных синдромом Дауна с тем его вариантом, для которого характерны транслокация и неполная трисомия 21, было обнаружено, что район хромосомы 21, управляющий экспрессией фациаль-ных, неврологических и сердечно-сосудистых изменений, ограничен пределами 21q22.2 и 21q22.3. Гены, располагающиеся внутри этого района, с которым связано развитие синдрома Дауна, должны играть решающую роль в патогенезе нарушений фенотипа.
Другие трисомии. Известны трисомии, затрагивающие хромосомы 8; 9; 22 и 13. По своей относительной частоте встречаемости заслуживают рассмотрения две трисомии — 18 и 13. Трисомия 18 (синдром Эдвардса; J.H.Edwards): задержка развития, слабоумие, глухота, парезы и параличи периферических нервов, маленькие челюсти, высокое небо, сдвоенная подковообразная почка и др.; частота — 1 наблюдение на 8000 родов; кариотипы 47,ХХ,+18 (рис. 8.6); тип мозаичности 46,ХХ/47,ХХ,+18. Трисомия 13 [синдром Патау, K.Patau; микроцефалия, слабоумие, заячья губа, волчья пасть (раздвоение верхней губы и верхнего неба) (рис. 8.7; 8.8), мор-
 |
Рис. 8.6. Кариотип 47, XX, + 18 (синдром Эдвардса). Окраска флюоро-хромом Hoechst 33258. Метафазная пластинка и раскладка хромосом.
щинистая кожа, аномалии почек и др.; частота — 1 наблюдение на 6000 родов; кариотип: трисомия тип 13 — 47,ХХ,+13; тип транслокации 46,XX,-14,+t(14q 13q); тип мозаичности — 46,ХХ/47,ХХ,+13].
В большинстве случаев обе трисомии развиваются в результате нерасхождения хромосом в мейозе, что приводит к носи-тельству излишней копии хромосомы 18 или 13. Как и при синдроме Дауна, отмечается связь с пожилым возрастом матери. Но в отличие от трисомии 21 в данном случае пороки развития гораздо тяжелее и многообразнее. Лишь немногие дети переживают 1-й год жизни, большинство погибают в течение нескольких месяцев после рождения.
 Синдром «кошачьего крика» (Cri du chat). Так было названо нарушение кариотипа, выражающееся в укорочении плеча хромосомы 5 (5р-). Детям в возрасте до 1 года, имеющим такой дефект, свойствен характерный плач, похожий на кошачье мяуканье. Важнейшие клинические признаки: тяжелая умственная отсталость, микроцефалия, круглая форма лица. Такие дети растут лучше, чем их сверстники с трисомиями, и некоторые доживают до зрелого возраста. По мере роста в периоде новорожденности они могут постепенно терять «кошачий крик» и высокий голосовой регистр.
Синдром «кошачьего крика» (Cri du chat). Так было названо нарушение кариотипа, выражающееся в укорочении плеча хромосомы 5 (5р-). Детям в возрасте до 1 года, имеющим такой дефект, свойствен характерный плач, похожий на кошачье мяуканье. Важнейшие клинические признаки: тяжелая умственная отсталость, микроцефалия, круглая форма лица. Такие дети растут лучше, чем их сверстники с трисомиями, и некоторые доживают до зрелого возраста. По мере роста в периоде новорожденности они могут постепенно терять «кошачий крик» и высокий голосовой регистр.
Цитогенетические заболевания, затрагивающие половые хромосомы. Эти болезни распространены гораздо шире, чем болезни, связанные с аутосомными аберрациями. Более того, несоответствие (избыток или трата) генетического материала в половых хромосомах переносится значительно лучше подобных несоответствий в аутосомах. Такая переносимость объясняется
Рис. 8.8. Синдром Патау: заячья губа и волчья пасть.
двумя факторами, характерными для половых хромосом: возможностью инактивации всех хромосом, кроме X, а также небольшим объемом генетического материала в хромосомах Y.
В 1961 г. Мэри Лайон (M.Lyon) охарактеризовала инактивацию хромосомы X и предположила то, что позднее стало широко известным как гипотеза Лайон. Было констатировано, что лишь одна из хромосом X является генетически активной, другая же независимо от того, имеет она отцовское или материнское происхождение, подвергается гетеропикнозу (иначе окрашивается) и остается неактивной. Инактивация происходит по случайному выбору в каких-либо клетках бластоцисты (зародышевого пузырька) примерно на 16-й день эмбриогенеза, в дальнейшем она постоянно сохраняется у той же хромосомы X в клетках-потомках каждого предшественника. Поэтому у большинства нормальных женщин обнаруживаются мозаичность и две клеточные популяции: одна с инактивированной материнской хромосомой X, другая с инактивированной отцовской хромосомой Y. В этом заключается объяснение того, почему у женщин имеется то же количество активных генов, относящихся к хромосоме X, что и у мужчин. Неактивная хромосома X хорошо определяется в интерфазном ядре в качестве интенсивно окра-
Схема 8.7. Кариотип (идиограмма) Х-хромосомы, полученная с помощью бендинга


Синдром тестикулярнои феминизации Х-сцепленный тяжелый иммунодефицит
Х-сцепленная агаммаглобупинемия Болезнь Фабри
Специфический ген-транскрипт Х-инактивации
Синдром Леша-Нихана
Гемофилия В Синдром Хантера Хрупкость Х-хромосомы Гемофилия А Недостаточность G6PD
Обозначения: Справа обозначения приблизительных локализаций важнейших генов (локу-сов) а также заболевания, возникающие при поражении X-хромосом Буквенные символы генов: POLA а-полимераза ДНК, DMD мышечная дистрофия Дюшенна, OTC — орнитинтранскарбами-паза, CGD хронический гранулематоз, TIMP тканевый ингибитор металлопротеиназ. AR рецептор андрогенов, PGK1 фосфоглицераткиназа 1, GLA а-гвлактозидаэа, HPRT гипоксантин-фосфорибоэилтрансфераза, G6PD глюкозо-6-фосфатдегидрогеназа; MIC2 белок клеточной поверхности, STS стероидсульфатаза, А1S9T фермент Е1, активирующий убиквитин, RPS4X — ри-босомный белок S4.
шенного и примыкающего к ядерной мембране тельца Барра (M.L.Barr), или полового хроматина, Х-хроматина. Половой хроматин имеется во всех соматических клетках нормальных женщин, но особенно хорошо он выявляется в мазках-отпечатках буккального эпителия слизистой оболочки щек.
Основные положения гипотезы Лайон выдержали проверку временем, но, разумеется, были уточнены. Например, вначале полагали, что все гены в неактивной хромосоме X находятся в выключенном состоянии. Затем выяснилось, что многие из них избегают инактивацию этой хромосомы (схема 8.7). Считается, что по крайней мере несколько генов, экспрессированных в обеих хромосомах X, важны для нормального роста и развития. Это подтверждается тем, что у больных с моносомией хромосомы X (симптом Тернера; 45,Х) ярко выражены тяжелые соматические и гонадные нарушения. Если одиночная «доза» (количество) Х-связанных генов достаточна, то никакого вредного эффекта нет. Более того, хотя одна из хромосом X во всех клетках инактивируется в ходе эмбриогенеза, она избирательно реактивируется в зародышевых клетках перед первым мейотическим делением. Таким образом, вероятно, обе хромосомы X нуждаются в нормальном овогенезе (развитии яйцеклеток).
Установлено, что ген, картированный в Xql3, служит в качестве главного переключателя, необходимого для подавления большинства генов в неактивной хромосоме X. Поразительно, но этот ген, названный геном Х-неактивного специфического транскрипта (XIST) (транскрипт — это продукт транскрипции), не кодирует синтез белка. Ясно, что РНК-транскрипт гена XIST никогда не покидает клеточного ядра и каким-то (пока необъяснимым) образом, предупреждает транскрипцию других генов в хромосоме X. Нарушения в гене XIST, предотвращающие нормальную инактивацию хромосомы X, могут объяснить, почему в определенных семьях встречается передача по женской линии Х-связанных особенностей, например гемофилии.
Хромосома Y определяет развитие мужского пола. Причем наличие одной хромосомы Y безотносительно от количества хромосом X уже детерминирует мужской пол. Ген, предопределяющий развитие яичек (Sry, или ген района Y, детерминирующего пол), локализуется в концевой зоне короткого плеча хромосомы Y.
Перечислим несколько признаков, общих для всех заболеваний, связанных с половыми хромосомами. Эти заболевания приводят к появлению ряда сложных и долговременных проблем при половом развитии, влияют на плодовитость. Часто эту патологию трудно распознать при рождении, и многие такие заболевания впервые диагностируют в период половой зрелости. Наконец, чем больше количество хромосом X у женщин или мужчин, тем выше вероятность возникновения умственной отсталости.
Синдром Клайнфелтера (H.F.Klinefelter), или мужской гипогонадизм (недоразвитие половых органов и вторичных половых признаков), встречается у лиц с двумя или более хромосомами X или двумя или более хромосомами Y. Среди генетических заболеваний, которые затрагивают половые хромосомы, этот синдром является одним из распространенных. Он же служит едва ли не самой частой причиной мужского гипогонадизма и встречается примерно у 1 из 850 живорожденных мальчиков. До наступления полового созревания синдром Клайнфелтера распознается редко, поскольку нарушения в яичках до ранних фаз полового созревания не проявляются. У большинства больных имеется ряд характерных признаков, например увеличение длины ног на всем протяжении между стопами и лонными костями, что создает впечатление удлиненного тела. Характерен евнухоидизм, при котором наряду с ненормально длинными ногами обнаруживаются маленькие, атрофичные яички, часто очень маленький половой член, а также отсутствие таких вторичных половых признаков, как низкий мужской голос, рост бороды и оволосение лобка по мужскому типу. Средний показатель умственного развития несколько ниже нормы, но умственная отсталость отмечается редко. Однако такой типичный вариант заболевания встречается не во всех случаях. Единственным постоянным признаком является гипо-гонадизм. Выявляется также стойкое повышение уровня плазменного гонадотропина, в частности фолликулостимулирующе-го гормона, в то время как количество тестостерона снижено. Увеличено содержание плазменного эстрадиола. В каждом отдельном случае степень феминизации определяется соотношением в плазме крови уровней эстрогенов и тестостерона.
Синдром Клайнфелтера — основная причина мужского бесплодия. Снижение сперматогенеза связано с несколькими видами морфологических изменений в яичках. У некоторых больных канальцы яичек полностью атрофированы и замещены нежным оксифильным гиалиновым и коллагеновым материалом. У других пациентов дисгенезия (нарушение эмбрионального развития) характеризуется гистологически нормальными канальцами, чередующимися с атрофичными. Иногда все канальцы яичек выглядят примитивными, как у эмбриона, и состоят из клеточных тяжей, в которых никогда не было просветов и которые не участвуют в развитом сперматогенезе. Обращают на себя внимание многочисленные клетки Лейдига (F.Leydig; гландулоциты яичка, находящиеся в стромальных перегородках), что обусловлено атрофией и скоплениями канальцев.
Классический вариант синдрома Клайнфелтера связан с кари-отипом 47,XXY (82 \% наблюдений). Такой набор формируется после нерасхождения хромосом во время мейотических делений у одного из родителей. Немногим более 50 \% таких наблюдений охватывает отцов; из оставшейся материнской части большинство случаев связано с нерасхождением хромосом при первом делении в мейозе. Между теми, кто получает лишнюю хромосому X от отца, и теми, кто получает ее от матери, нет никакой фенотипи-ческой разницы. В случаях, связанных с ошибками в овогенезе, возраст матери обычно немолодой. Кроме указанного классического варианта кариотипа, примерно у 15 \% больных обнаруживают мозаичность набора, чаще выражающуюся как 46,XY/47,XXY. Бывают и другие разновидности кариотипа: 47,XXY/48,XXXY, изредка даже 48,XXXY или 49.XXXXY. У таких лиц с полисомией X обнаруживаются всевозможные физические нарушения: криптор-хизм (задержка одного или обоих яичек в забрюшинном пространстве и неопущение их в мошонку), гипоспадия (аномалия или отсутствие какой-либо части уретры), более тяжелые формы недоразвития яичек, а также изменения скелета, например про-гнатия (выступающая верхняя челюсть) и радиоульнарный синостоз (сращение).
Синдром XYY. Дополнительные хромосомы Y могут быть обнаружены у мужчин с наборами 47,XYY или даже больше, вплоть до полисомии Y. Примерно 1 из 1000 живорожденных мальчиков имеет какой-либо из этих кариотипов. Почти все мальчики с такими измененными наборами фенотипически нормальны, однако для них характерны слишком большой рост и особенно высокая частота возникновения кожных угрей в тяжелой форме (угри — воспаление сальных желез и волосяных фолликулов). Умственное развитие нормальное.
Трактовка влияния лишней хромосомы Y на поведение больных остается спорной. Повышенная частота такого карио-типа наблюдается у заключенных в тюрьму уголовных преступников. Отмечают поведенческие отклонения по антисоциальному типу (без склонности к насилию) правонарушителя. Последние исследования показали, что лишь 1—2 \% лиц с кариотипом XYY имеют аномальное поведение. Подавляющее большинство из них — не более антисоциальны, чем другие люди с меньшим количеством хромосом Y.
Синдром Тернера (H.H.Turner) развивается при полной или частичной моносомии хромосомы X и характеризуется прежде всего гипогонадизмом у лиц с фенотипически женским полом. Встречается с частотой 1 на 3000 новорожденных девочек. Примерно у 57 \% больных полностью отсутствует одна хромосома X и имеется классический кариотип 45,X. У остальных 43 \% возникают другие аберрации. Следует отметить, что лишь около 1 \% плодов с кариотипом 45,Х доживают до рождения. У выживших новорожденных обнаруживаются тяжелые повреждения. В отличие от других форм анэуплоидии половых хромосом вариант 45,Х синдрома Тернера часто распознают уже при рождении или в раннем детстве.
Более 50 \% пациенток с указанным синдромом, которые не обладают кариотипом 45,Х, имеют структурные нарушения в хромосоме X. В порядке убывания частоты эти нарушения можно расположить следующим образом: делеция короткого плеча и формирование изохромосомы длинного плеча — 45,X,i (Xq); делеции частей короткого или длинного плеча — 46,XXq" или 4б,ХХр"; делеция частей и короткого, и длинного плечей с формированием кольцевой хромосомы — 4б,Х,г(Х). У оставшейся части больных обнаруживается мозаичность набора с такими кариотипами, как 45,X/46,XY или 45,Х/47,ХХХ. При синдроме Тернера важно оценить гетерогенность кариотипа, поскольку она связана со значительным разнообразием фенотипа. В отличие от больных с моносомией X, лица, имеющие мозаичность или делеции, например 46,XXq", могут обладать почти нормальной наружностью и жаловаться разве что на первичную аменорею (отсутствие менструаций).
У наиболее тяжело пораженных новорожденных, как правило, обнаруживают отек тыла кистей и стоп, вызванный лимфо-
14. Пальцев, т. 1.
417
стазом, и иногда опухание задней части шеи. Последнее связано с заметным расширением лимфатических сосудов, создающих так называемую кистозную гигрому (ограниченное скопление жидкости в ткани). По мере развития таких новорожденных опухание уменьшается, часто оставляя после себя двусторонний сетчатый рисунок на шее и стойкую дряблость кожи выи (шеи сзади). Наблюдаются и врожденные поражения сердца и крупных сосудов, особенно коарктация аорты (сужение в области дуги, стеноз устья аорты с эндокардиальным фиброэластозом. Все эти аномалии могут приводить к ранней гибели.
Важнейшие клинические признаки в юношеском и взрослом возрасте: маленький рост, опущение задней границы волосистой части головы, широкая грудная клетка и смещенные латерально соски молочных желез, cubitus valgus (деформированный или изогнутый, иногда кнаружи, локтевой сустав), недоразвитые яичники (в виде узких полосок), аменорея (отсутствие менструаций), бесплодие, множество пигментных невусов. В пубертатном периоде не развиваются вторичные половые признаки (по R.S.Cotran, V.Kumar, T.Collins, 1998). Гениталии остаются инфантильными, молочные железы не достигают адекватного развития, волосистость лобковой зоны небольшая. Ментальный статус у таких больных обычно нормален, лишь некоторые из них страдают умственной неполноценностью. Из всех перечисленных признаков для распознавания патологии у взрослых больных особую важность имеют очень маленький рост, редко превышающий 150 см, и аменорея. Синдром Тернера — одна из наиболее важных причин первичной аменореи (около 30 \% наблюдений).
В ходе овогенеза обе хромосомы X активны и необходимы для нормального развития яичников. Чтобы лучше осмыслить патогенез синдрома Тернера, важно вспомнить некоторые детали его развития. Первоначально яичники у плодов содержат до 7-Ю6 овоцитов. Последние начинают исчезать еще при внутриутробном развитии, а к моменту рождения из 7106 овоцитов остается около 4-Ю6. Перед наступлением менархе (первой менструации) их число уже не превышает 400 ООО. Дальнейшая утрата овоцитов продолжается в период половой зрелости, а когда наступает менопауза (прекращаются менструальный цикл и детородная функция), овоцитов остается менее 10 000.
При синдроме Тернера яичники плода развиваются нормально. Однако отсутствие второй хромосомы X приводит к ускоренной потере овоцитов, полная утрата которых наступает в 2-летнем возрасте — менопауза опережает менархе. Яичники атрофируются до узких фиброзных полосок и лишаются яйцеклеток и фолликулов. Уменьшенная продукция эстрогенов сопровождается повышенной выработкой гонадотропина (суммарное название гормонов передней доли гипофиза, стимулирующих формирование и активность половых желез).
Рис. 8.9. Двурогая матка.
Множественные хромосомы X. Встречаются кариотипы с 1—3 лишними хромосомами X, их частота составляет 1 на 1200 новорожденных. Большинство женщин с такими кариотипами полностью нормальны. Однако встречаются и разнообразные случайные находки. Как уже отмечалось, тенденция к умственной отсталости возрастает пропорционально количеству лишних хромосом X. Поэтому слабоумие наблюдается у всех женщин с кариотипом 49,ХХХХХ, в то время как большинство лиц с кариотипом 47,ХХХ не поражены. У некоторых женщин бывает аменорея или нарушения менструального цикла.
Гермафродитизм и ложный гермафродитизм. Проблема половой неопределенности исключительно сложна. Пол индивидуума определяется на нескольких уровнях. Генетический пол детерминирован (обусловлен) наличием или отсутствием хромосомы Y. Неважно, сколько при этом имеется хромосом X, одна хромосома Y уже определяет развитие яичек и мужской генетический пол. Первоначально индифферентные гонады и мужских, и женских эмбрионов имеют свойственную им тенденцию феминизироваться до тех пор, пока не начнут действовать маскулинизирующие Y-хромосомозависимые факторы. Гонадный пол обусловлен анатомо-гистологическими характеристиками половых желез. Дуктальный пол зависит от наличия дериватов парамезонефрального протока (мюллеров про-
14*
419
ток, J.P.Mueller; из этого канала образуется эпителий матки, маточных труб и влагалища, у мужчин он редуцируется в простатическую маточку) (рис. 8.9) или мезонефрального протока (вольфов проток, C.F.Wolff, из него образуется семявыносящий проток, у женщин он редуцируется в рудимент внутри придатка яичника). Фенотипический, или генитальный, пол зависит от строения наружных половых органов. Если между перечисленными признаками имеется «несогласованность», то возникает половая неопределенность.
Термин «истинный гермафродитизм» подразумевает одновременное наличие яичниковой и тестикулярной тканей. В отличие от этого при ложном гермафродитизме имеется «несогласованность» между фенотипическим и гонадным полами. Иными словами, человек с женским ложным гермафродитизмом имеет яичники, но мужские наружные гениталии, а с мужским ложным гермафродитизмом — тестикулярную ткань, но гениталии женского типа.
Истинный гермафродитизм встречается редко. У некоторых людей обнаруживают яичко на одной стороне и яичник на другой. В то же время бывают и комбинации из овариотестикуляр-ной ткани, называемые ovotestes. У 2А больных кариотип 4б,ХХ, у большинства остальных имеется мозаичность, например XX/XXY, при которой обнаруживается линия клеток—носителей хромосомы Y. При кариотипе 4б,ХХ происходит транслокация хромосомы Y на хромосому X или аутосому, а при мозаичности имеются хромосомы X и Y. Таким образом, люди с истинным гермафродитизмом составляют разнородную группу, чаще всего имея две хромосомы X и полную либо неполную хромосому Y по крайней мере в некоторых клетках.
Женский ложный гермафродитизм представляется гораздо менее сложным. Во всех случаях генетический пол выражен как XX, и развитие! гонад (яичников) и внутренних половых органов происходит нормально. Только наружные половые органы неопределенны или вирилизованы (развиты по мужскому типу). Это заболевание обусловлено чрезмерной и неадекватной подверженностью плода действию андрогенных гормонов в раннем периоде внутриутробного развития. Эти стероиды чаще всего продуцируются надпочечниками плода, пораженными наследственной гиперплазией, передающейся как аутосомно-рецессивное заболевание. В этих случаях возникают дефекты в синтезе гидрокортизона (кортизола), которые вторично приводят к избыточному синтезу андрогенных гормонов корой надпочечников плода.
Мужской ложный гермафродитизм — наиболее сложный вариант из всех половых нарушений генетической природы. Больные обладают хромосомой Y, поэтому их гонады представлены исключительно яичками. Однако половые пути в наружных половых органах дифференцированы по мужскому фенотипу неполностью. Эти гениталии или «неопределенны», или полное тью феминизованы (развиты по женскому типу). Мужской ложный гермафродитизм очень неоднороден и имеет множество причин. Общей для каждого случая является дефектная вирилизация мужского эмбриона, которая происходит в результате генетически детерминированных нарушений в синтезе и/или действии андрогенов. Наиболее распространенная форма, названная синдромом полной андрогенной нечувствительности, или тес-тикулярной феминизации, развивается после мутаций гена рецептора андрогенов. Этот ген локализуется в зоне Xqll— Xql2, поэтому данное заболевание наследуется как Х-связанное рецессивное.

Обсуждение Патологическая анатомия
Комментарии, рецензии и отзывы